DOI:
10.1039/C0PY00147C
(Paper)
Polym. Chem., 2010,
1, 1453-1458
Received
8th May 2010
, Accepted 8th June 2010
First published on
11th August 2010
Abstract
The amphiphilic azobenzene block copolymer, poly(6-(4-phenylazophenoxy)hexylmethacrylate-b-poly((2-dimethylamino)ethyl methacrylate)) (P(PHMA)-b-P(PEGMA)) was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization technique. The results showed that the polymerization was carried out in a controlled manner. The obtained amphiphilic block copolymer showed strong fluorescence emission in mixed water/tetrahydrofuran (THF) solution. The intensity of fluorescence emission was slightly changed after the solution was irradiated by UV-vis light. Further investigation showed that the copolymer emitted strong fluorescence in THF after UV-vis irradiation. The polymer solution was characterized by DLS and TEM. The results showed that the change of fluorescence emission behavior of the amphiphilic block copolymer both in water–THF and THF solution was caused by the formation and size change of aggregates originating from the photo-isomerization of azobenzene chromophore.
Introduction
Azobenzene compounds were well known for their unique reversible trans-cis-trans isomerization. These compounds were widely used as photo-responsive materials,1–4 such as optical data storage, liquid crystal displays, optical switching, holographic surface relief gratings (SRGs). Recently, the photo-induced fluorescence emission was found to be a unique property of azobenzene compounds.5–12 Some possible reasons were proposed in different systems, such as acidification of azobenzene chromophores,5,6 the formation of bilayer structures7,8 or even the photo induced self-assembly of azobenzene moieties.9,10 Bo and Zhao first expanded the fluorescent azobenzene research into the azobenzene-containing diblock copolymer system.11 An amphiphilic block copolymer with pendant azobenzene hydrophobic and quaternized poly(4-vinyl pyridine) hydrophilic structure showed enhanced fluorescence emission after adding different amounts of water into the polymer/DMF solution. They ascribed the fluorescence behavior to the subtle change in the state of polymer micellar association, which may alter the confining state of azobenzene groups. Following this work, our group found similar results for azobenzene-containing block copolymers with the poly((2-dimethylamino)ethyl methacrylate) hydrophilic chains.12 Furthermore, our more recent work showed that the azobenzene end-functionalized polystyrene (PS) self-assembled to nano scale aggregation in CHCl3 under the UV-irradiation.13 This unique self-assembly behavior was attributed to the polarity change of the azobenzene isomerized from its trans form to cis form after UV-irradiation, which showed different solubility in CHCl3.14 The formed aggregation showed strong fluorescence emission from azobenzene chromophore.
Above reports indicated that the azobenzene chlormophore can show fluorescence emission in some conditions, such as the formation of micelle or aggregation. But the mechanism of fluorescent emission by azobenzene chromophore is not yet clearly understood. Thus, it is necessary to further explore fluorescent azobenzene-containing diblock copolymers system with a deep investigation of the cause of fluorescence behavior. Amphiphilic diblock copolymers prefer to form micelles or aggregates through self-assembly in selective solvents.15 Many kinds of azobenzene containing amphiphilic diblock copolymers already have been prepared.16–19 Their photo responsive self-assembly behavior has been explored at the same time. Among them, block copolymers with poly(ethylene glycol) (PEG) as the hydrophilic fragment shows some advances, such as insensitivity to pH and good biocompatibility etc. Amphiphilic azobenzene-containing block copolymer with PEG as hydrophilic fragment was reported by Wang et al.19 The micelle formed by self-assembly of this copolymer showed interesting photo-induced shape deformation under UV-irradiation.
In this paper, we selected poly(ethylene glycol) methyl ether acrylate (PEGMA) as the building block to synthesize azobenzene containing diblock copolymers. The PEGMA segment was the alternative selection with similar properties to PEG.20 Furthermore, the PEGMA segment can be easily introduced into the block copolymer chain by well-developed living free radical polymerization (LFRP) techniques, such as atom transfer radical polymerization (ATRP)21 and reversible addition fragmentation chain transfer (RAFT)22 polymerization. Here, we synthesized the amphiphilic azobenzene-containing polymer, poly(6-(4-phenylazophenoxy)hexylmethacrylate-b-poly((2-dimethylamino)ethyl methacrylate)) (P(PHMA)-b-P(PEGMA)), via RAFT method. The fluorescence behavior of the obtained copolymer was investigated in water–THF and THF solution, respectively.
Experimental
Materials
Poly(ethylene glycol) methyl ether acrylate (PEGMA) (Mn = 475 Da, Aldrich) was purified by passing through a basic alumina column to remove the inhibitor. 2,2′-Azobisisobutyronitrile (AIBN, Shanghai Chemical Reagent Co., China) was recrystallized twice from ethanol solution. Tetrahydrofuran (THF, Shanghai Chemical Reagent Co., China) and anisole (analytical reagent, Shanghai Chemical Reagent Co., China) were purified according to the standard procedure. 2-Cyanoprop-2-yl-1-dithionaphthalate (CPDN)23 and 6-(4-phenylazophenoxy)hexylmethacrylate (PHMA)24 were synthesized according to the literature, respectively. 1-Chloro-6-hydroxyhexane (Alfa Aesar, 95%) and other reagents were all used as received without any further purification.
PHMA (2.74 g, 7.50 mmol), AIBN (12.3 mg, 0.075 mmol), and CPDN (61.4 mg, 0.225 mmol) were dissolved in 10 mL of anisole in a 25 mL flask. After purging with argon for 10 min to eliminate the oxygen, the flask was sealed under argon atmosphere and placed in an oil bath held by a thermostat at the desired temperature (80 °C) to polymerize for 4 h. At the end of the reaction, the flask was cooled in ice water then opened. The contents were dissolved in 20 mL of THF, precipitated into 400 mL of methanol. The polymer (poly(PHMA), P(PHMA)) was obtained by filtration and then purified by reprecipitating twice from THF to methanol and dried in vacuum overnight at 40 °C. The conversion of polymerization was 52.3% as determined gravimetrically.
Preparation of amphiphilic diblock copolymer P(PHMA)-b-P(PEGMA)s
The P(PHMA)-b-P(PEGMA) block copolymers were prepared using similar procedures to that of RAFT polymerization of PHMA, except that PPHMA was used as the macro-RAFT agent. The typical experiment was carried out as follows: a mixture of PEGMA (0.475 g, 1 mmol), PPHMA18 (Mn(GPC) = 6900 g mol−1, Mw/Mn = 1.20, 138 mg, 0.02 mmol), AIBN (0.65 mg, 0.004 mmol) ([PEGMA]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[AIBN]0:[macro-RAFT]0 = 250:1:5), and 3 mL of anisole were added into a 5 mL ampoule. The solution was purged with argon for 10 min to eliminate the oxygen. Then, the ampoule was flame-sealed and placed in an oil bath held by a thermostat to polymerize at 70 °C. At timed intervals, the tube was cooled in ice water then opened. The contents were dissolved in 10 mL of THF, precipitated into 250 mL of petroleum ether. The polymers obtained were dried in vacuum at room temperature to a constant weight.
:[AIBN]0:[macro-RAFT]0 = 250:1:5), and 3 mL of anisole were added into a 5 mL ampoule. The solution was purged with argon for 10 min to eliminate the oxygen. Then, the ampoule was flame-sealed and placed in an oil bath held by a thermostat to polymerize at 70 °C. At timed intervals, the tube was cooled in ice water then opened. The contents were dissolved in 10 mL of THF, precipitated into 250 mL of petroleum ether. The polymers obtained were dried in vacuum at room temperature to a constant weight.
Preparation of the micelle solutions
The diblock copolymers P(PHMA)-b-P(PEGMA) were first dissolved in anhydrous THF (a good solvent for both blocks) to obtain solutions with concentrations of 0.5 mg mL−1. Then the different volumes of Milli-Q water were added into the prepared polymer solutions at a rate of one drop every 10 s by a microsyringe under vigorous stirring, respectively. After the water addition was completed, another 9 mL of water was added to the dispersions to quench the formed colloidal structures. Stable dispersions obtained by removing THF at room temperature under a dust-free air ambience condition for 48 h.
Analysis and characterizations
The number average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of the polymers were determined using a Waters 1515 gel permeation chromatographer (GPC) equipped with a refractive index detector, using HR1 (pore size: 100 Å, 100–5000 Da), HR2 (pore size: 500 Å, 500–20 000 Da), and HR4 (pore size 10 000 Å, 50–100 000 Da) columns (7.8 × 300 mm, 5 μm beads size) with a molecular weight range of 100–500,000 Daltons, calibrated with poly(methyl methacrylate) (PMMA) standard samples. THF was used as the eluent at a flow rate of 1.0 mL min−1 operated at 30 °C. GPC samples were injected using a Waters 717 plus autosampler. 1H-NMR spectra were recorded on an INOVA 400 MHz nuclear magnetic resonance (NMR) instrument, using CDCl3 as a solvent, tetramethylsilane (TMS) as the internal standard. Ultraviolet visible (UV-vis) absorption spectra of micelle solutions in selective solvents were performed on a Shimadzu (Kyoto, Japan) UV-240. The fluorescence emission spectra of the polymers were obtained on a PerkinElmer LS-50B fluorescence spectrophotometer. Transmission electron microscopy (TEM) was recorded on a Tecnai G2–20 TEM at a 200 kV accelerating voltage. The samples were prepared by mounting a drop of the micelle solution (0.05 mL) on a copper EM grid covered with a thin film of formvar. The spherical aggregate size analysis was performed using dynamic light scattering (DLS) measurements (Zetasizer Nano ZS: Malvern Instrument Ltd. UK) at 20 °C.
Results and discussion
The RAFT polymerization technique is widely used to prepare polymers with pre-determined structures. Azobenzene-containing polymers can be synthesized in a controlled manner using this technique.12 Here, we reported the synthesis of diblock copolymer containing PEGMA and azobenzene polymer segments. The synthetic route to the diblock copolymer of PHMA and PEGMA is shown in Scheme 1. Firstly, the azobenzene homopolymer, poly(6-(4-(phenylazo)phenoxy)hexyl methacrylate) (PPHMA), was synthesized by RAFT polymerization using 2-cyanoprop-2-yl-1-dithionaphthalate (CPDN) as the RAFT agent and 2,2′-azobisisobutyronitrile (AIBN) as the initiator at 80 °C. The PPHMA was obtained with a conversion of 52.3% after 4 h polymerization. The obtained PPHMA had a molecular weight of 6900 g mol−1 and a molecular weight distribution of 1.20. The repeat unit number of the polymer can be calculated viaeqn (1), where the numbers of 6900, 271 and 366 are the molecular weight of polymer, CPDN and PHMA respectively. The result showed that the obtained polymers contain 18 unit of monomer. This polymer labeled as PPHMA18. Then, using PPHMA18 as the macro-RAFT agent, PEGMA was polymerized in anisole at 70 °C using AIBN as the initiator.| |
| Repeat Number = (6900 − 271)/366 ≈ 18 | (1) |
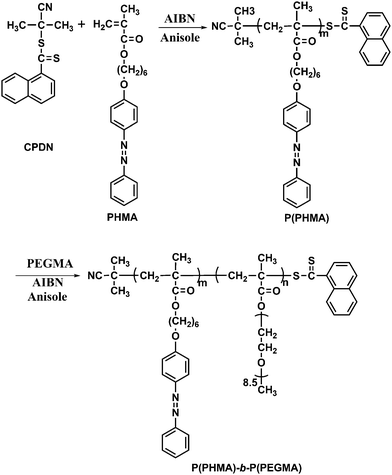
Amphiphilic diblock copolymers, P(PHMA)-b-P(PEGMA)s, with different molecular weight are successfully prepared using PPHMA18 as the macro-RAFT agent with the molar ratio of [PEGMA]0:[AIBN]0:[macro-RAFT]0 = 250:1:5. As shown in Fig. 1A, the linear relationship between ln([M]0/[M]) and the polymerization time for the RAFT polymerization of PEGMA indicated that the propagating radical concentration remains constant during the process of polymerization. Fig. 1B shows the plots of number-average molecular weight measured by GPC (Mn(GPC)) versus the PEGMA conversion, along with the corresponding change in their molecular weight distribution (Mw/Mn). A linear fitting of data can be observed for the diblock copolymers with relatively narrow molecular weight distribution (Mw/Mn < 1.25), which is evidence of controlled growth of the PEGMA block and living nature of the polymerization. Moreover, at the molar feed ratio of [PEGMA]0:[AIBN]0:[PPPHM]0 = 250:1:5, the evolution of GPC curves as a function of reaction time (Fig. S1†) clearly indicates a controlled growth of the second PEGMA block via RAFT polymerization. Table 1 lists the characteristics of the diblock copolymers. The compositions of the block polymer are obtained as follows: the Mn(GPC) of the azo homopolymers is used to calculate the number of monomeric units of the first block, and the number of the second PEGMA block is calculated according to the composition of the diblock copolymers as revealed by their 1H NMR spectra, respectively. Fig. 2 shows the typical 1H NMR spectra of P(PHMA)18 and P(PHMA)18-b-P(PEGMA) in CDCl3. The relative amounts of phenyl protons of azobenzene (d in Fig. 2) and PEGMA groups (I in Fig. 2) can be easily obtained by comparing the integration values of these peaks. The molecular weights of the block copolymers can be estimated according to the compositions determined by 1H NMR spectra (eqn (2)). The results are summarized in Table 1. It can be seen that the Mn(GPC) is much smaller than that of Mn(NMR) for each sample, which may be due to the difference of hydrodynamic volumes between the block copolymers and the linear PMMA standards.25 Despite the uncertainty about the absolute number of monomeric units, the composition of the block copolymer as revealed by 1H NMR gives the relative length of the two blocks.
| |
| Mn(NMR) = ((I3.2–3.4/3)/(I6.8–7.0/2))×18 × Mn, PEGMA + 6900 | (2) |
| Sample |
Time/h |
Conv. (%) |
M
n(GPC)
|
M
n(NMR)
|
M
w/Mnd |
|
The polymerization was carried out in anisole at 70 °C with the reaction ratio of [monomer]0):[AIBN]0:[Macro-RAFT]0 = 250:1:5, PPHMA18 was prepared via RAFT polymerization using CPDN as the RAFT agent.
Number average molecular weight measured by GPC using PMMA as the standard.
Number average molecular weight measured by 1H NMR according to eqn (2).
Molecular weight distribution measured by GPC.
|
| PPHMA18 |
— |
— |
6900 |
— |
1.20 |
| P(PHMA)18-b-P(PEGMA)4 |
3 |
7.8 |
7000 |
8800 |
1.21 |
| P(PHMA)18-b-P(PEGMA)5 |
5 |
20.3 |
7400 |
9300 |
1.24 |
| P(PHMA)18-b-P(PEGMA)14 |
7 |
32.7 |
8800 |
13500 |
1.24 |
| P(PHMA)18-b-P(PEGMA)22 |
9 |
56.9 |
11100 |
17400 |
1.21 |
| P(PHMA)18-b-P(PEGMA)29 |
14 |
80.8 |
12200 |
20700 |
1.24 |
![The dependence of ln([M]0/[M]) on the polymerization time (h) (A) and molecular weights and molecular weight distribution (Mw/Mn) on the conversion (B) for the RAFT polymerization of PEGMA. Polymerization conditions: [PEGMA]0 : [AIBN]0 : [macro-RAFT]0 = 250 : 1 : 5, T = 70 °C, [PEGMA]0 = 3.3 mol L−1, anisole = 3 mL.](/image/article/2010/PY/c0py00147c/c0py00147c-f1.gif) |
| | Fig. 1 The dependence of ln([M]0/[M]) on the polymerization time (h) (A) and molecular weights and molecular weight distribution (Mw/Mn) on the conversion (B) for the RAFT polymerization of PEGMA. Polymerization conditions: [PEGMA]0:[AIBN]0:[macro-RAFT]0 = 250:1:5, T = 70 °C, [PEGMA]0 = 3.3 mol L−1, anisole = 3 mL. | |
 |
| | Fig. 2
1H NMR spectra of P(PHMA)18 and P(PHMA)18-b-P(PEGMA) 14 in CDCl3 with TMS as internal standard. | |
Where, I3.2–3.4 and I6.8−7.0 are the integrals of 3.2–3.4 ppm and 6.8–7.0 ppm, respectively. The numbers 3, 2, 18 and 6900 are proton number in i, proton number in d, the repeat unit of P(PHMA) and molecular weight of P(PHMA), respectively.
Fluorescence property of PPHMA-b-P(PEGMA)
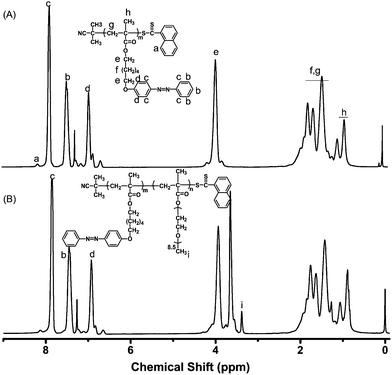
Some researchers reported that the azobenzene polymers showed fluorescence emission in some cases.5–12 The intensity of the fluorescence emission is sensitive to UV irradiation. Most of these polymers have amphiphilic structures. Here, we investigated the fluorescence property of the obtained azobenzene-containing amphiphilic diblock copolymer. The results presented in Fig. 3 show that these copolymers solution emit strong fluorescence centered at 409 nm in water–THF system after excited by 320 nm light. The fluorescence quantum yield was estimated as 0.049 using a similar method to that in ref. 26. Normally, the azobenzene compound is nonfluoresent due to the fast nonradiative relaxation process of fast and high efficient tans-cis isomerization for excited azobenzene.11 However, this fast relaxation process can be restricted by packing the azobenzene moieties into nano-scale aggregations, which encourages excited azobenzene moieties to return to their ground state via the radiative process, e.g. through fluorescent emission. The formation of the nano-scale aggregations was verified by the TEM images as given in Fig. 4. The amphiphilic diblock copolymer self assembled to uniform nano-scale aggregations in water–THF solution.
 |
| | Fig. 3 Fluorescent emission spectra of P(PHMA)18-b-P(PEGMA) 14 in water–THF solution before and after irradiation at 365 nm for different time. The exciting wavelength (λex) is 320 nm. The concentration is 1.5 × 10−5 mol L−1. | |
 |
| | Fig. 4 TEM images of the micellar aggregates of P(PHMA)18-b-P(PEGMA) 14 in H2O–THF solution. A: before UV-irradiation; B: after 30 min UV-irradiation. | |
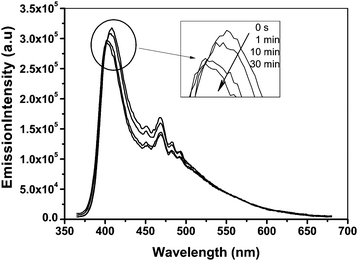
The results in Fig. 3 showed that the fluorescence emission intensity of P(PHMA)18-b-P(PEGMA)14 in water–THF solution can be effected by UV-irradiation. The fluorescence emission intensity slowly decreases with UV-irradiation time. The maximum wavelength of emission also slowly shifted to a short number. Both of them are reached their stable value after UV-irradiation of 30 min. These results are contrary to the results previously reported by Han et al.10 and ourselves.13 Although the exact reason for the UV-irradiation enhance fluorescence emission is far from being understood, these early reported cases indicated that the change of aggregate size should be responsible for this enhancement.10,13 The fluorescent intensity seems to be directly proportional to its aggregate size. Fig. 4B indicated that the aggregation size of P(PHMA)-b-P(PEGMA) in water–THF solution is slightly decreased from ∼38 nm to ∼30 nm after 30 min of 365 nm light irradiation. This result is confirmed by DLS. As shown in Fig. 5, the aggregation size decreased from 92 nm to 85 nm after it was irradiated by 365 nm UV for 30 min. Thus, the decrease in the aggregation diameter results in the change of fluorescence emission. The exact reason still is unclear. However, it shows that the decrease of aggregation size reduces the percentage of exited azobenzene relaxation by radiative way, which evidently resulted in a decrease of fluorescence emission intensity and blue-shift of maximum value. It should be noted that the difference between the diameter of aggregation obtained by TEM and DLS should be the result of with and without solvent in the determination status.
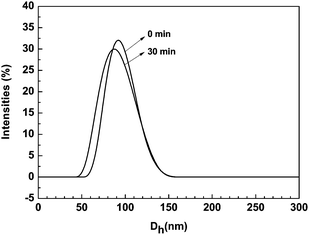 |
| | Fig. 5 Size distribution for P(PHMA)18-b-P(PEGMA) 14 in water–THF solution before and after irradiating with 365 nm light determined by DLS. The concentration is 1.5 × 10−5 mol L−1. | |
However, the sensitivity of fluorescence emission with UV-irradiation shows very weak in the water–THF system. It has been reported that the cis form azobenzene showed larger polarity than that of the trans form,14 which caused changes in the hydrophilic-hydrophobic balance of P(PHMA)-b-P(PEGMA). Thus, more hydrophilic polymer was formed due to the azobenzene moiety isomerized to its cis form after UV-irradiation, which may cause changes in aggregation diameter. However, the aggregation structure hindered the isomerization of azobenzene (low isomerization rate is founded in aggregation system, Fig. 6), which also weakened the size change of the aggregation before and after UV-irradiation. Thus, low sensitivity of fluorescence emission with UV-irradiation is observed in current system.
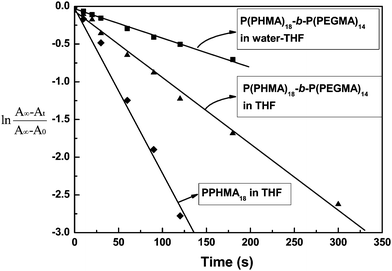 |
| | Fig. 6 First-order plots for trans-cis isomerization of P(PHMA)18 in THF and P(PHMA)18-b-P(PEGMA) 14 in water–THF and THF solutions. The concentrations are both 1.5 × 10−5 mol L−1. | |
The kinetic of azobenzene moiety trans-cis isomerization was investigated to help better understand the fluorescence emission process. The results are shown in Fig. 6. It indicated that the isomerization rate for the azobenzene in water–THF system (rate constant k ≈ 3.89 × 10−3 s−1) was significantly smaller than that in THF (rate constant k ≈ 5.83 × 10−3 s−1). This result confirmed that the trans-cis isomerization of azobenzene was hindered after the formation of aggregation, which may favor fluorescence emission. At the same time, the results in Fig. 6 also show that the isomerization rate of P(PHMA)18-b-P(PEGMA)14 was slower than normal azobenzene polymers, even for the amphiphilic sample.11 These results encouraged us to investigate the fluorescence emission behavior of current block copolymer in THF, which was considered to be a good solvent for both chain segments in P(PHMA)-b-P(PEGMA) block copolymer. The results are shown in Fig. 7.
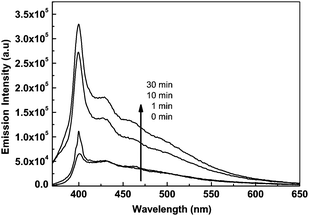 |
| | Fig. 7 The dependence of fluorescent emission intensity of P(PHMA)18-b-P(PEGMA) 14 in THF solution on time of irradiation at 365 nm. The exciting wavelength (λex) is 320 nm. The concentration is 1.5 × 10−5 mol L−1. | |
The results in Fig. 7 show that the P(PHMA)-b-P(PEGMA) block copolymer solution showed weak fluorescence emission at 400 nm before it was irradiated by 365 nm light. The intensity of this fluorescence peak significantly increased after the polymer solution was irradiated by 365 nm light. It reached the saturation state after 30 min of irradiation. The fluorescence quantum yields before and after 30 min of UV light irradiation were estimated as 0.003 and 0.046 respectively according to the method in ref. 26. The results showed that the enhancement of fluorescence emission by UV-light irradiation in THF solution was more sensitive than that in water–THF system. Moreover, the fluorescent emission intensity was increased with the time of 365 nm light irradiation in THF, which showed the opposite effect to that in the water–THF system. It should be noted that very weak fluorescence emission (can be neglected compare to the block copolymer) can be observed in the THF solution of PPHMA homopolymer even after the irradiation of 365 nm light (ESI, Fig. S4†). In order to find out the reason for this, DLS was used to track the change in the block copolymer solution before and after the irradiation of 365 nm light. The results shown in Fig. 8 indicate that nano-scale aggregation was formed in the solution after irradiation of 365 nm light. Thus, this aggregation will hinder excited azobenzene relaxation in a nonradiative way, which resulted in the fluorescence emission. Furthermore, the diameter of the aggregates increased with the irradiation time. Similar results were found in azobenzene terminated polystyrene system in our previous report.13 The increase of aggregation size resulted in the enhancement of fluorescence emission. Although the actual reason for the formation of aggregates after UV-irradiation still is unclear, it can be speculated that the trans-cis isomerization of azobenzene should be the reason for both the irradiation induced aggregation and size change. The polarity change after azobenzene isomerized from its trans form to cis form caused the solubility change of the azobenzene moiety in THF, which was the origin for the formation of aggregates.13 However, the introduction of the P(PEGMA) segment was necessary for the formation of aggregation in current case, because no aggregation is found for the homopolymer of P(PHMA) in the same conditions.
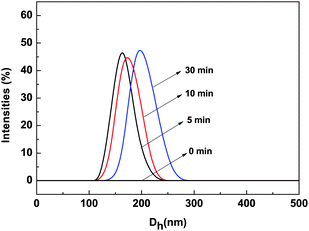 |
| | Fig. 8 Size distribution of P(PHMA)18-b-P(PEGMA)14 in THF before and after irradiating at 365 nm for different lengths of time determined by DLS. The concentration is 1.5 × 10−5 mol L−1. | |
Conclusion
An amphiphilic block copolymer of P(PHMA)-b-P(PEGMA) was successfully synthesized in a controlled manner via the RAFT technique. The chain segment ratio can be adjusted via conversion. The obtained block copolymers showed fluorescence emission centered at 409 nm after micellar aggregation in water–THF solution due to the hindered nonradiative trans-cis isomerization relaxation of azobenzene moiety. The fluorescence emission intensity was slightly decreased with the time of 365 nm irradiation accompanied with a weakly decrease in the aggregation size. The trans-cis isomerization rate of copolymer (5.83 × 10−3 s−1) was slower than that of P(PHMA) homopolymer (2.25 × 10−2 s−1) even in THF solution. The THF solution of P(PHMA)-b-P(PEGMA) showed intense fluorescence emission at 400 nm after the irradiation of 365 nm light. The intensity of this emission is dramatically enhanced with irradiation time. The similar nano-scale aggregation was observed in the THF solution of copolymer after irradiation, which was due to the polarity change after azobenzene isomerized from trans to cis form. The introduce of P(PEGMA) segment into azobenzene was necessary to form the nano-scale aggregation. The size of such nano-scale aggregation was increased from zero to 160 nm and 200 nm after 5 min and 30 min of irradiation, respectively.
Acknowledgements
The financial support of this work by the National Natural Science Foundation of China (Nos. 20874069, 50803044), the Specialized Research Fund for the Doctoral Program of Higher Education contract grant (No. 200802850005), the Program of Innovative Research Team of Soochow University and the Qing Lan Project are gratefully acknowledged.
Notes and references
-
(a) G. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915 CrossRef CAS;
(b) D. Brown, A. Natansohn and P. Rochon, Macromolecules, 1995, 28, 6116 CrossRef CAS;
(c) S. J. Ziker, T. Bieringer, S. Haarer, R. S. Stein, J. W. van Egmond and S. G. Kostromine, Adv. Mater., 1998, 10, 855 CrossRef CAS;
(d) A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139 CrossRef CAS;
(e) J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817 CrossRef CAS;
(f) C. J. Barrett, J. Mamiya, K. G. Yagerc and T. Ikeda, Soft Matter, 2007, 3, 1249 RSC;
(g) Y. Zhao and J. He, Soft Matter, 2009, 5, 2686 RSC.
-
(a) W. M. Gibbons, P. J. Shannon, S. T. Sun and B. J. Swetlin, Nature, 1991, 351, 49 CrossRef CAS;
(b) T. G. Pedersen, P. M. Johansen and H. C. Pedersen, J. Opt. A: Pure Appl. Opt., 2000, 2, 272 CrossRef CAS;
(c) D. Hore, A. Natansohn and P. Rochon, J. Phys. Chem. B, 2003, 107, 2197 CrossRef CAS;
(d) H. R. Hafiz and F. Nakanishi, Nanotechnology, 2003, 14, 649 CrossRef CAS;
(e) P. H. Rasmussen, P. S. Ramanujam, S. Hvilsted and R. H. Berg, J. Am. Chem. Soc., 1999, 121, 4738 CrossRef CAS.
-
(a) E. Ishow, B. Lebon, Y. N. He, X. G. Wang, L. Bouteiller, L. Galmiche and K. Nakatani, Chem. Mater., 2006, 18, 1261 CrossRef CAS;
(b) B. L. Lachut, S. A. Maier, H. A. Atwater, M. J. A. de Dood, A. Polman, R. Hagen and S. Kostromine, Adv. Mater., 2004, 16, 1746 CrossRef;
(c) Q. Zeng, Z. A. Li, Z. Li, C. Ye, J. G. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634 CrossRef CAS;
(d) H. Yu, A. Shishido, Y. Iyoda and T. Ikeda, Macromol. Rapid Commun., 2007, 28, 927 CrossRef CAS.
-
(a) X. B. Chen, Y. H. Zhang, B. J. Liu, J. J. Zhang, H. Wang, W. Y. Zhang, Q. D. Chen, S. H. Pei and Z. H. Jiang, J. Mater. Chem., 2008, 18, 5019 RSC;
(b) W. H. Li, S. Nagano and T. Seki, New J. Chem., 2009, 33, 1343 RSC;
(c) D. R. Wang, G. Ye, Y. Zhu and X. G. Wang, Macromolecules, 2009, 42, 2651 CrossRef CAS;
(d) Q. Kulikovska, L. M. Goldenberg, L. Kulikovsky and J. Stumpe, Chem. Mater., 2008, 20, 3528 CrossRef CAS;
(e) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2010, 1, 78 RSC.
- C. H. Tung and J. Q. Guan, J. Org. Chem., 1996, 61, 9417 CrossRef CAS.
- Z. Lei, A. Vaidyalingam and P. K. Dutta, J. Phys. Chem. B, 1998, 102, 8557 CrossRef CAS.
- M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175 CrossRef CAS.
- K. Tsuda, G. C. Dol, T. Gensch, J. Hofkens, L. Latterini, J. W. Weener, E. W. Meijer and F. C. Schryver, J. Am. Chem. Soc., 2000, 122, 3445 CrossRef CAS.
- M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951 CrossRef CAS.
- M. Han, Y. Hirayama and M. Hara, Chem. Mater., 2006, 18, 2784 CrossRef CAS.
- Q. Bo and Y. Zhao, Langmuir, 2007, 23, 5746 CrossRef.
- J. Xu, W. Zhang, N. Zhou, J. Zhu, Z. Cheng, Y. Xu and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5652 CrossRef CAS.
- F. Ma, N. Zhou, J. Zhu, W. Zhang, L. Fan and X. Zhu, Eur. Polym. J., 2009, 45, 2131 CrossRef CAS.
-
Y. Zhao and T. Ikeda, Smart Light-Responsive Materials: Azobenzene-Containing Polymers and Liquid Crystals, Wiley, New Jersey, 2009, p. 215 Search PubMed.
- M. Moffitt, K. Khougaz and A. Eisenberg, Acc. Chem. Res., 1996, 29, 95 CrossRef CAS.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911 CrossRef CAS.
- L. Ding, H. Mao, J. Xu, J. He, X. Ding, T. P. Russell, D. R. Robello and M. Mis, Macromolecules, 2008, 41, 1897 CrossRef CAS.
- W. Su, H. Zhao, Z. Wang, Y. Li and Q. Zhang, Eur. Polym. J., 2007, 43, 657 CrossRef CAS.
- D. Wang, G. Ye, Y. Zhu and X. Wang, Macromolecules, 2009, 42, 2651 CrossRef CAS.
- O. G. Schramm, G. M. Pavlov, H. P. Erp, M. A. R. Meier, R. Hoogenboom and U. S. Schubert, Macromolecules, 2009, 42, 1808 CrossRef CAS.
-
(a) J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901 CrossRef CAS;
(b) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS;
(c) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS;
(d) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
-
(a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery and P. T. Le, Macromolecules, 1998, 31, 5559 CrossRef CAS;
(b) Y. K. Chong, P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071 CrossRef CAS;
(c) A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS;
(d) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS;
(e) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- J. Zhu, X. L. Zhu, Z. P. Cheng and F. Liu, Polymer, 2002, 43, 7037 CrossRef CAS.
- H. Ringsdorf and H. W. Schmidt, Makromol. Chem., 1984, 185, 1327 CAS.
- Z. P. Cheng, X. L. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21, 7180 CrossRef CAS.
- Y. Pang, J. Li and T. J. Barton, J. Mater. Chem., 1998, 8, 1687 RSC.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.