Versatile post-polymerization functionalization of poly(p-phenylene vinylene) copolymers containing carboxylic acid substituents: development of a universal method towards functional conjugated copolymers†
Jan
Duchateau
a,
Laurence
Lutsen
b,
Wanda
Guedens
a,
Thomas J.
Cleij
*a and
Dirk
Vanderzande
ab
aHasselt University, Institute for Materials Research (IMO), Agoralaan, Building D, B-3590, Diepenbeek, Belgium. E-mail: thomas.cleij@uhasselt.be; Fax: +32-11-268301; Tel: +32-11-268310
bIMEC, Division IMOMEC, Wetenschapspark 1, B-3590, Diepenbeek, Belgium
First published on 7th June 2010
Abstract
A novel conjugated copolymer of poly(2-methoxy-5-(3,7-dimethyloctyloxy)-1,4-phenylene vinylene) (MDMO-PPV) and poly(1,4-(2-(5-carboxypentyloxy)-5-methoxyphenylene) vinylene) (CPM-PPV), which can be used as a platform for further post-polymerization functionalization, is presented. This copolymer is soluble in common organic solvents, such as CHCl3, CH2Cl2 and THF. Post-polymerization functionalization of the acid-groups of this copolymer has been successfully performed via an optimized DCC/DMAP-procedure with the desired alcohol. Several examples have been prepared to demonstrate the versatility of the developed functionalization methodology. PPV-copolymers have been prepared with vinyl groups, propynylphenyl groups, methacrylate groups, ATRP initiator groups, dithiocarbamate initiator groups and propargyl groups. The latter group can be utilized to do “click” chemistry, which has been demonstrated with an azide-functionalized reagent. In all cases, analytical data are consistent with a quantitative conversion of the functional groups. It is demonstrated that no substantial degradation of the conjugated system occurs even after consecutive post-polymerization functionalization reactions.
Introduction
Conjugated polymers are unique materials, which combine the electronic and optical properties of semiconductors with the mechanical properties and processability of polymers. As a result, conjugated polymers are useful for a wide range of electronic applications. Polymeric materials have the advantage of being lightweight, flexible, low-cost and straightforward to process. In addition, the diversity of possible synthetic methods can lead to an infinite variability of compounds, which allows for the tailoring of the optical and electronic properties for a specific application. Among the numerous applications of conjugated polymers are polymer light emitting diodes,1–4 field effect transistors,5–7 (bio)sensors8–10 and photovoltaic devices.11–13Numerous methods can be found in the literature for the actual tailoring process of conjugated polymers.14–18 Hitherto, a significant drawback is the lack of a universal approach, leading to a wide variety of methods and functionalization procedures to obtain specific functional materials. To avoid this drawback, we have developed a universal platform and methodology, which allow for further functionalizations in a uniform manner. Within the wide range of available conjugated polymers, our focus is on the conjugated polymer poly(p-phenylene vinylene) (PPV) and its derivatives. Notwithstanding, a similar approach can be utilized for other conjugated polymers. As a platform for further functionalization, the copolymer 8 (MDMO-CPM)-PPV, has been developed (Scheme 1). This is a copolymer of poly(2-methoxy-5-(3,7-dimethyloctyloxy)-1,4-phenylene vinylene) and poly(1,4-(2-(5-carboxypentyloxy)-5-methoxyphenylene) vinylene. The side-chains of (MDMO-CPM)-PPV consist of branched C10-alkylchains and alkyl chains with carboxylic acid groups in a ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This copolymer is prepared via the sulfinyl precursor route19,20 and post-functionalization of the carboxylic acid groups is done via an optimized DCC/DMAP-method with the desired alcohol.
:1. This copolymer is prepared via the sulfinyl precursor route19,20 and post-functionalization of the carboxylic acid groups is done via an optimized DCC/DMAP-method with the desired alcohol.
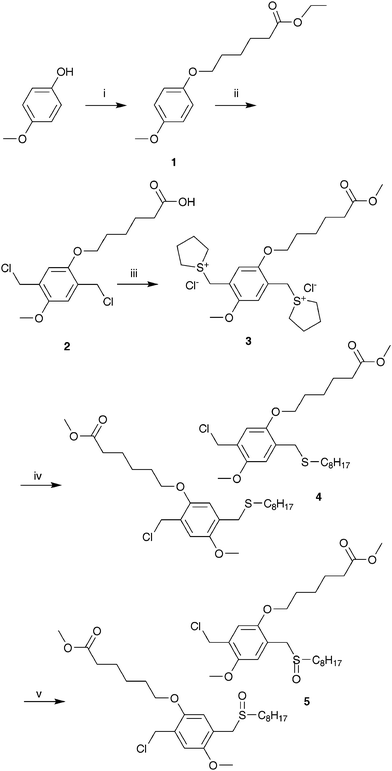 | ||
| Scheme 1 Synthesis of the functional sulfinyl monomer 5 (i: NaOtBu, ethyl 6-bromohexanoate, NaIcat, EtOH (reflux); ii: p-CH2O, Ac2O, HCl, 60 °C; iii: THT, MeOH, 50 °C; iv: C8H17SH, NaOtBu, MeOH; v: H2O2, TeO2, HClcat, 1,4-dioxane). | ||
This article describes the synthesis of the carboxylic acid containing copolymer (MDMO-CPM)-PPV 8 (n/m = 9) and the model compound 6-(4-methoxy-phenoxy)-hexanoic acid 9. The carboxylic acid groups of 9 react readily with alcohol functionalized molecules via a DCC/DMAP-method to give the corresponding carboxylic esters. The optimized DCC/DMAP-procedure is translated to the copolymer 8, giving identical excellent results as observed for model compound 9. Using this method seven novel functionalized PPV derivatives have been prepared. This demonstrates the versatility of both our conjugated polymer based platform as well as the developed universal functionalization methodology. It is now possible to post-polymerization functionalize PPV derivatives, with virtually any functional group, which allows for the development of many new and exciting applications.
Experimental section
General
NMR spectra were recorded with a Varian Inova Spectrometer (1H-NMR 300 MHz, 13C-NMR 75 MHz). Analytical size exclusion chromatography (SEC) was performed using a Spectra series P100 (Spectra Physics) pump equipped with a pre-column (5 μm, 50 × 7.5 mm, guard, Polymer Labs) and two mixed-B columns (10 μm, 2 × 300 × 7.5 mm, Polymer Labs) and a refractive index detector (Shodex) at 40 °C. THF was used as the eluent at a flow rate of 1.0 mL min−1. Molecular weight distributions are given relative to polystyrene standards. Gas chromatography/mass spectrometry (GC-MS) analyses were carried out on TSQ–70 and Voyager mass spectrometers (Thermoquest); the capillary column was a Chrompack Cpsil5CB or Cpsil8CB. DIP-MS measurements were performed at a heating rate of 10 °C min−1 up to 600 °C. In this technique the material is brought directly on the heating element of the probe as a thin film and this enables to study the products that are liberated in the high vacuum (4 × 10−4 Pa) inside the spectrometer. Liberated products were ionized by electron impact. FT-IR spectra were collected with a Perkin Elmer Spectrum One FT-IR spectrometer (nominal resolution 4 cm−1, summation of 16 scans). UV-Vis measurements were performed on a Cary 500 UV-Vis-NIR spectrophotometer (scan rate 600 nm min−1, continuous run from 200 to 800 nm). Unless stated otherwise, all reagents and chemicals were obtained from commercial sources and used without further purification. PMDETA was distilled under vacuum. Cu(I)Br was purified by stirring in acetic acid for 24 h, to reduce Cu(II) to Cu(I). Subsequently, Cu(I)Br was filtered off and washed with ethanol and diethyl ether to remove Cu(II)Br2 and acetic acid, after which it was dried in a vacuum oven at 75 °C.Monomer synthesis
The functional monomer 5, i.e. the mixture of 6-(5-chloromethyl-4-methoxy-2-octylsulfinylmethyl-phenoxy)-hexanoic acid methyl ester and 6-(2-chloromethyl-4-methoxy-5-octylsulfinylmethyl-phenoxy)-hexanoic acid methyl ester, as well as all intermediates 1, 2, 3 and 4 were prepared via a modified literature procedure (Scheme 1).15 Experimental details can be found in the ESI.† The sulfinyl monomer for MDMO-PPV (Scheme 2) was prepared according to a literature procedure.21 All properties were identical to the previously reported material. The synthesis of model compounds 9–17 (Scheme 3), which were used for reaction optimizations, can be found in the ESI as well.†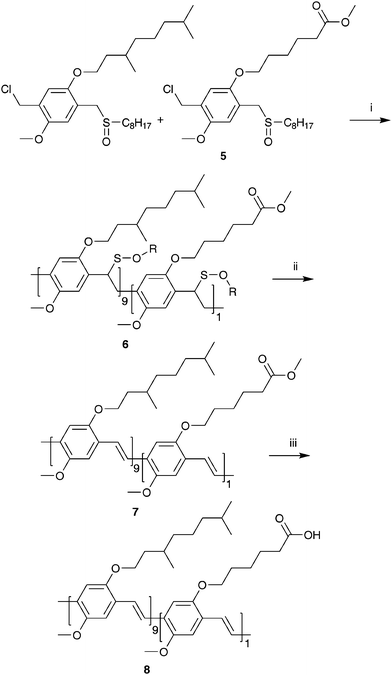 | ||
| Scheme 2 Synthesis of the copolymers 6, 7 and 8 (i: NaOtBu, 2-BuOH, 30 °C; ii: toluene; iii: KOtBu, H2O, 1,4-dioxane (reflux)). The synthesis is shown for only one set of regio-isomeric monomers. | ||
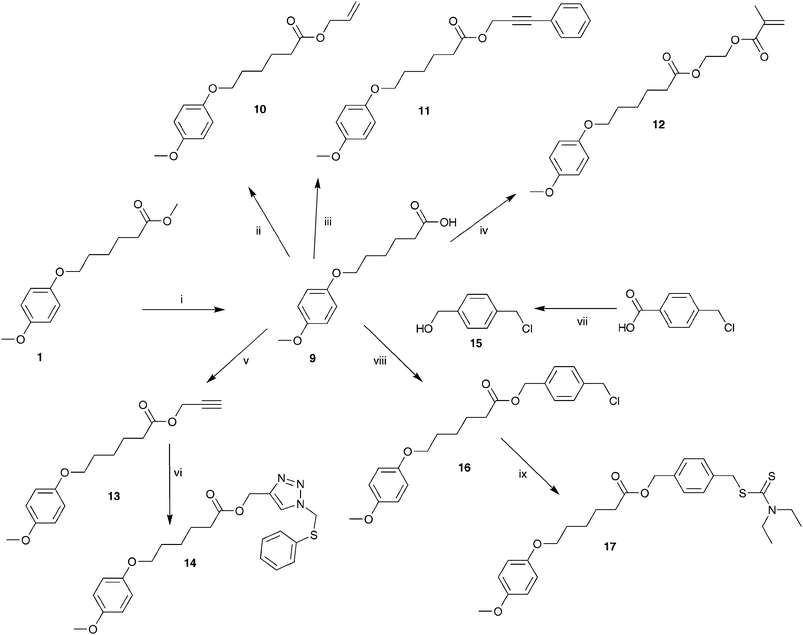 | ||
| Scheme 3 Synthesis of model compound 9 and further functionalizations to give compounds 10–17 (i: KOtBu, H2O, MeOH, 50 °C; ii: DCC, DMAP, CH2Cl2, allylalcohol; iii: DCC, DMAP, CH2Cl2, 3-phenyl-2-propyn-1-ol; iv: DCC, DMAP, CH2Cl2, 2-hydroxyethyl-methacrylate; v: DCC, DMAP, CH2Cl2, propargylalcohol; vi: PMDETA, Cu(I)Br, azidomethyl phenyl sulfide, THF; vii: BH3·THF, THF; viii: DCC, DMAP, CH2Cl2; ix: sodium diethyldithiocarbamate trihydrate, THF). | ||
Precursor copolymer 6 towards (MDMO-CPM)-PPV
The molar monomer feed ratio of the copolymerization reaction was 1 equivalent of sulfinyl monomer 5 to 9 equivalents of the MDMO sulfinyl monomer. A thermostatic flask (30 °C) was charged with a mixture of sulfinyl monomer 5 (0.047 g, 0.1 mmol) and the MDMO sulfinyl monomer (0.438 g, 0.9 mmol) in 2-BuOH (30 mL). NaOtBu (0.125 g, 1.3 mmol), dissolved in the same solvent (20 mL), was added in one portion via a thermostatic funnel after both solutions were purged with N2. Polymerization was allowed to proceed for 1 h at 30 °C. The reaction was terminated by pouring the reaction mixture in a well stirred amount of ice water (250 mL). After extraction with CH2Cl2 (3 × 150 mL), the combined organic layers were evaporated under reduced pressure giving the crude precursor copolymer 6, which was used without further purification. 1H-NMR (CDCl3): δ = 7.0–6.3, 4.5, 4.1–3.5, 3.4, 2.5–2.2, 1.9–0.6; FT-IR (NaCl, cm−1): 2954, 2922, 2856, 1730 (νC–O), 1505, 1464, 1408, 1384, 1366, 1327, 1261, 1217, 1044, 870; SEC (THF) Mw = 150 × 103 g mol−1 (PD = Mw/Mn = 3.6). UV-Vis λmax = 301 nm (THF).Ester-functionalized copolymer 7
A stirred solution of copolymer 6 (0.475 g) in toluene (50 mL) was purged with N2 for 30 min, after which the elimination reaction was allowed to proceed at 110 °C and stirred for 3 h.Subsequently, toluene was evaporated under reduced pressure and 7 was redissolved in THF (30 mL). The resulting red solution was precipitated drop wise in cold MeOH (400 mL). The resulting red copolymer was filtered off, washed with cold MeOH and redissolved in toluene (50 mL). The elimination procedure was repeated after which pure 7 was filtered off, washed with cold MeOH and dried at room temperature under reduced pressure as a red, fibrous polymer (0.244 g, 85% yield). 1H-NMR (CDCl3): δ = 7.6–7.4, 7.2–7.1, 4.1–3.8, 3.63, 2.4–2.3, 1.9–0.6; FT-IR (NaCl, cm−1): 2958, 2922, 2851, 1733 (νC–O), 1504, 1464, 1413, 1384, 1353, 1259, 1205, 1094, 1030, 968, 860, 800; SEC (THF) Mw = 540 × 103 g mol−1 (PD = Mw/Mn = 2.5). UV-Vis λmax = 510 nm (THF).
(MDMO-CPM)-PPV 8
A solution of copolymer 7 (0.244 g, 0.085 mmol ester functionalities) and dioxane (50 mL) was heated to reflux temperature after which a solution of KOtBu (0,094 g, 0.85 mmol) in water (1 mL) was added at reflux temperature. After 4 h stirring at reflux temperature, CH2Cl2 (150 mL) was added to the reaction mixture and extracted with 1 N HCl-solution (100 mL). The organic layer was evaporated under reduced pressure giving copolymer 8. The resulting red copolymer was dried at room temperature under reduced pressure (0.230 g, 95% yield). 1H-NMR (CDCl3): δ = 7.6–7.4, 7.2–7.1, 4.1–3.8, 2.4–2.3, 1.9–0.6; FT-IR (NaCl, cm−1): 3059, 2954, 2923, 2855, 1709 (νC–O), 1505, 1464, 1414, 1383, 1353, 1259, 1205, 1095, 1035, 968, 857, 801; SEC (THF) Mw = 270 × 103 g mol−1 (PD = Mw/Mn = 3.2). UV-Vis λmax = 507 nm (THF).Post-polymerization functionalization (Scheme 4)
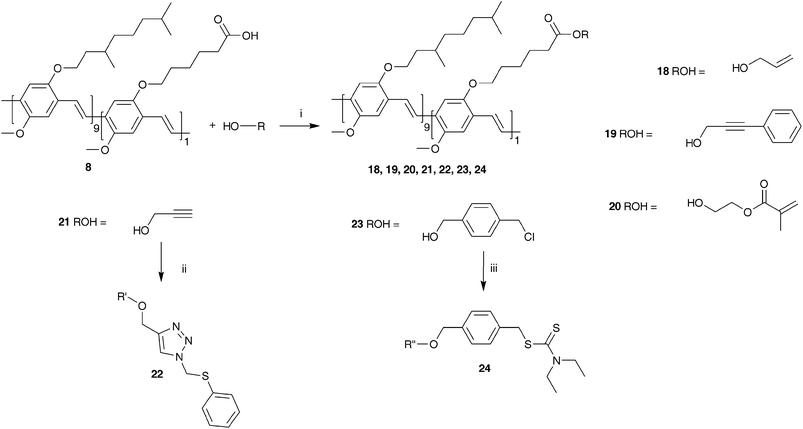 | ||
| Scheme 4 Synthesis of copolymers 18–24via the optimized DCC/DMAP-procedure (i: DCC, DMAP, CH2Cl2; ii: PMDETA, Cu(I)Br, azidomethyl phenyl sulfide, THF; iii: sodium diethyldithiocarbamate trihydrate, THF). | ||
Results and discussion
Monomer synthesis (Scheme 1)
To obtain high chemical selectivity during polymerization, the sulfinyl route was chosen as the precursor method for the desired PPV derivatives.19–22 The use of precursor routes not only offers improved processing properties, but it also has the potential to result in devices with a better stability due to the possibility of removing part of the solubilizing groups, i.e. the dithiocarbamate groups, from the final conjugated polymer by post polymerization elimination. Such post polymerization processing procedures of conjugated polymer films are highly promising for device applications also have advantages for the ultimate device stability, which is of crucial importance in organic electronics.23–26 To obtain optimal solubility, a copolymer was synthesized starting from the MDMO sulfinyl monomer21 and monomer 5, both of which actually are a mixture of two regio-isomers, i.e. 6-(5-chloromethyl-4-methoxy-2-octylsulfinylmethyl-phenoxy)-hexanoic acid methyl ester and 6-(2-chloromethyl-4-methoxy-5-octylsulfinylmethyl-phenoxy)-hexanoic acid methyl ester.15 However, the mixture of region-isomers is used without further separation as monomers for the polymerization. The resulting absence of regioregularity in the corresponding conjugated polymers is typical for PPV-type polymers prepared via precursor routes. Only by an elaborate separation of the 1/1 mixture of regio-isomers of the sulfinyl monomers, regioregular PPV-derivatives can be obtained.27 Monomer 5 was prepared in a five step reaction according to Scheme 1. Starting with a Williamson etherification between 4-methoxyphenol and ethyl-6-bromohexanoate, ester 1 was obtained, which was chloromethylated using concentrated HCl and formaldehyde in acetic anhydride giving 2. Subsequently, the bissulfonium salt 3 was synthesized by stirring 2 and THT in MeOH. The mono-substituted thio-ether 4 was formed by reaction of the symmetrical bissulfonium salt 3 with an equimolar amount of an alkylthiolate anion and afterwards azeotropic removal of tetrahydrothiophene. Finally, selective mono-oxidation gave the desired sulfinyl monomer 5.Copolymer synthesis (Scheme 2)
The desired copolymer is composed of the well known MDMO sulfinyl monomer and the sulfinyl monomer 5 in a feed-ratio of 9/1. The actual built in ratio can be determined via1H-NMR spectroscopy. The signals at 0.99 ppm are assigned to the CHCH3 groups in the MDMO part of the copolymer. The triplet at 2.35 ppm originates from the CH2COOCH3 in the ester part of the copolymer. It is found that the ratio of the two comonomers built in is circa 9% ester monomer and circa 91% MDMO-monomer, which is in good agreement with the feed-ratio. This copolymer has an optimal composition to obtain sufficient solubility in a variety of common organic solvents. A higher built-in ratio of carboxylic acid groups results in a reduced solubility, which can hamper further functionalization in a reproducible manner. The polymerization reaction of the sulfinyl monomers proceeds via a p-xylylene intermediate, i.e. a p-quinodimethane system, which is the actual monomer in the formation of the precursor copolymer 6.Thermal conversion of the precursor copolymer 6 gave the conjugated copolymer 7. This conversion is readily visible with solution UV-Vis spectroscopy. Whereas the λmax of the precursor copolymer 6 is 301 nm, the corresponding conjugated copolymer 7 has a clearly red shifted λmax at 510 nm (Table 1, Fig. 1). This elimination process of the sulfinyl groups of 6 is monitored by in situ UV-Vis spectroscopy using a dynamic heating program of 2 °C min−1 from ambient temperature up to 146 °C under a continuous N2 flow (Fig. 2). At room temperature, the thin film of 6 exhibits a strong absorbance with a maximum at λmax = 301 nm. Upon heating, a new absorption band appears related to the conjugated system. At increasing temperatures, this band exhibits a gradual red shift (λmax = 469 nm at 110 °C). After cooling down to room temperature, λmax is even more red shifted to 510 nm as a result of a reversible thermochromic effect.22 When the absorbance at this maximum wavelength is monitored versus temperature, it becomes evident that the conjugated structure starts to develop around 75 °C (Fig. 2).
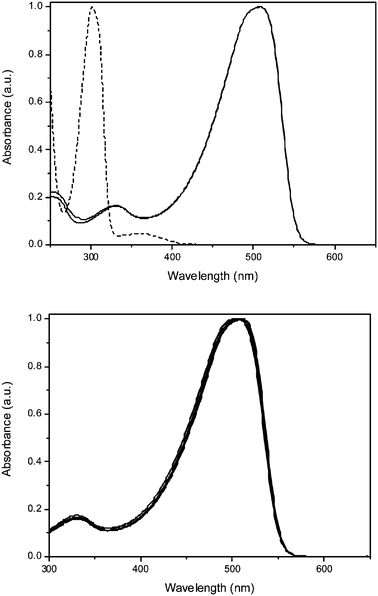 | ||
Fig. 1 Solution UV-Vis absorption spectra of all copolymers in THF. Top: copolymers 6 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), 7 ( ), 7 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and 8 () (the spectra of 7 and 8 are virtually indistinguishable from each other). Bottom: copolymers 8, 18, 19, 20, 21, 22, 23 and 24 () (no significant changes are observed upon functionalization). ) and 8 () (the spectra of 7 and 8 are virtually indistinguishable from each other). Bottom: copolymers 8, 18, 19, 20, 21, 22, 23 and 24 () (no significant changes are observed upon functionalization). | ||
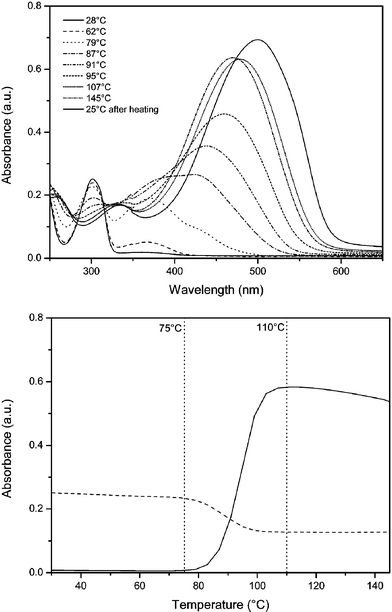 | ||
| Fig. 2 Thin film UV-Vis absorption spectroscopy of the gradual formation of the conjugated copolymer 7. Top: individual absorption spectra at selected temperatures. Bottom: absorbance at 301 nm () and 510 nm () as a function of temperature. | ||
The observed molecular weight, Mw, for precursor copolymer 6 is 1.5 × 105 g mol−1. The corresponding conjugated copolymer 7 has a somewhat higher Mw of 5.4 × 105 g mol−1 (Table 1). This apparent increase in molecular weight measured in THF solution using polystyrene standards can be explained by the significant differences in hydrodynamic volume and polarity of the conjugated copolymer, as compared to the precursor copolymer. In reality, it can be assumed that only a small decrease of the molecular weight occurs upon formation of the conjugated structure due to the elimination of sulfinyl groups. Notwithstanding, the observed Mw values confirm that high molecular weight polymers are obtained.
Hydrolysis of the ester group of copolymer 7 gave copolymer 8 in excellent yield. The almost quantitative conversion could be observed using IR spectroscopy, in which the νC–O of the ester at 1733 cm−1 was shifted to a value of 1709 cm−1 for the corresponding carboxylic acid (Fig. 3). The UV-Vis absorption spectrum of copolymer 8 is almost identical to that of copolymer 7 (Table 1, Fig. 1), indicating that no significant degradation of the conjugated system occurs upon hydrolyzing the ester functionalities. The Mw value for 8 is 2.7 × 105 g mol−1, which is also in the same order of magnitude as copolymer 7 (Table 1).
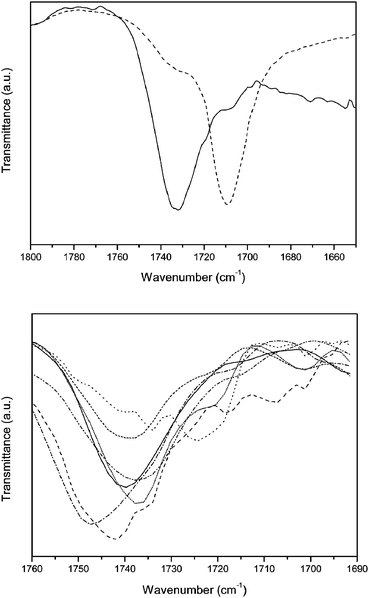 | ||
| Fig. 3 Thin film FT-IR spectroscopy of the conjugated polymers. Top: copolymers 7 (solid) and 8 (dashed). Bottom: copolymers 18 (solid), 19 (dash), 20 (dot), 21 (dash dot), 22 (dash dot dot), 23 (short dash) and 24 (short dot). | ||
To obtain more information about the thermal stability of the conjugated system of 8, an in situ UV-Vis heating experiment was performed under N2 atmosphere. To this end, a thin film of 8 was heated using a dynamic heating program at 2 °C min−1 from ambient temperature up to 120 °C and subsequently cooled down to room temperature. In the next run, the sample was heated to 140 °C and subsequently cooled down. This sequence was repeated up to 240 °C (Fig. 4). After each heating ramp, the film of 8 was cooled down to 25 °C and a UV-Vis absorption spectrum was taken at that temperature (Fig. 4). The absorption maximum measured of 8 in film at room temperature is λmax = 512 nm. By heating 8 the effective conjugation length decreases e.g. when 8 is heated up to 120 °C, the λmax is shifted from 512 nm to 496 nm. However, after subsequent cooling to 25 °C, the conjugated system is fully recovered and again a λmax of 512 nm is observed. This thermochromic effect can also be observed during the heating at higher temperatures. Up to temperatures of 220 °C, the original λmax is fully recovered upon cooling to 25 °C. In contrast, after heating at a temperature of 240 °C, a small blue-shift (λmax = 510 nm) is observed after cooling to 25 °C. This demonstrates that the heating process is no longer reversible, indicating that the onset of degradation of the conjugated system occurs around this temperature. Apparently, the effect on the thermal stability of the 9% built in carboxylic acid functionalizations is negligible, since a similar stability is observed as was found for MDMO-PPV.28 In conclusion, copolymer 8 is an excellent platform for further functionalization steps. It is soluble in common organic solvents like CHCl3, CH2Cl2 and THF. Post-functionalization of the carboxylic acid groups of copolymer 8 can therefore be done via an optimized DCC/DMAP-method with the desired alcohol.
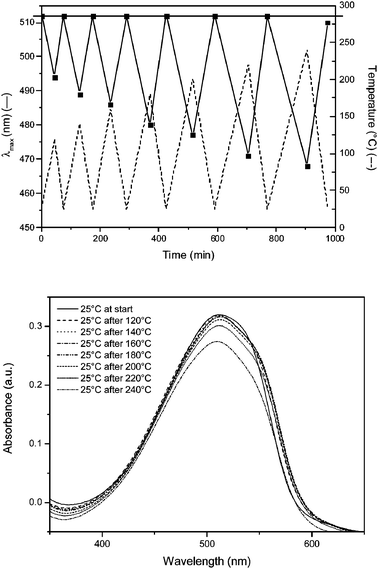 | ||
| Fig. 4 UV-Vis thermal stability measurement of conjugated polymer 8. Top: dynamic heating program () and absorption maximum as a function of time and temperature (). Bottom: individual thin film UV-Vis absorption spectra of 8 at room temperature after using a dynamic heating program. | ||
Model compound synthesis and functionalization
With copolymer 8 being readily available, post-polymerization functionalizations become possible. However, since the optimization of the reaction conditions on a conjugated polymer is not straightforward, due to synthetic and analytical limitations, first a model compound was prepared on which the reaction conditions were optimized (Scheme 3). Model compound 9 was obtained via a simple hydrolysis of the ester group of 1. The carboxylic acid function of 9 readily reacts with alcohol functionalized molecules via the optimized DCC/DMAP-method to the corresponding esters. This was tested with five hydroxyl-bearing compounds giving 10, 11, 12, 13 and 16. Alcohol 15 was synthesized by a reduction of 4-chloromethyl benzoic acid with BH3·THF. The propargyl group of 13 can be used to attach a desired molecule, with an azide group, via “click” chemistry. This was tested with an azide-functionalized reagent, i.e. azidomethyl phenyl sulfide, to form compound 14 with the help of PMDETA and Cu(I)Br. Compound 16 was also further functionalized to form compound 17, containing a dithiocarbamate group. Compounds 9, 10, 11, 12, 13, 14, 15, 16 and 17 were all characterized with 1H-NMR, 13C-NMR, FT-IR and mass spectroscopy and were pure. Our post-polymerization functionalization strategy has been exemplified by five groups of reactions.Copolymer functionalization
The quality of the conjugated system is further confirmed via UV-Vis measurements in THF. As can be seen in Table 1, the peak positions (λmax) of the π–π* transitions of the post-functionalized copolymers 18–23 are all positioned between 503 and 511 nm. This is the same spectral region as the transition observed for copolymer 8 (λmax = 507 nm). In addition to the peak positions, the shapes of the UV-Vis absorption spectra are also identical (Fig. 1). Apparently, the post-polymerization procedures have no significant impact on the quality of the conjugated system, further confirming the suitability of our approach as a general method towards advanced functional conjugated polymers.
Conclusions
In conclusion, a new copolymer (MDMO-CPM)-PPV 8 has been developed, which can be used as a platform for further post-polymerization functionalization. Copolymer 8 was prepared via the sulfinyl precursor route from the MDMO sulfinyl monomer as well as an ester functional monomer 5 in a ratio of 9/1. After a double thermal conversion of the precursor copolymer 6, the conjugated copolymer 7 was hydrolyzed to copolymer 8. This copolymer is soluble in common organic solvents, such as CHCl3, CH2Cl2 and THF. It has been fully characterized using different analytical techniques. Post-functionalization of the acid-group of copolymer 8 is done via an optimized DCC/DMAP-procedure with the desired alcohol. This procedure was first optimized and tested for model compounds, after which it was utilized to functionalize copolymer 8. In this way, copolymer 8 was post-functionalized with a variety of functional groups, which can be regarded as representative examples. PPV-copolymers were prepared with vinyl groups, propynylphenyl groups, methacrylate groups, ATRP initiator groups, dithiocarbamate initiator groups and propargyl groups. The latter group can be utilized to do “click” chemistry, which was demonstrated with an azide-functionalized reagent, i.e. azidomethyl phenyl sulfide. In all cases, FT-IR and 1H-NMR are consistent with a quantitative conversion of the functional groups. Furthermore, spectroscopic data and GPC measurements indicate that no substantial degradation of the conjugated system occurs even after consecutive post-polymerization functionalization reactions. This demonstrates the versatility of the developed universal functionalization methodology, which is not only of interest to functionalize PPV-type materials, but can also be utilized on other conjugated polymer systems.Acknowledgements
The authors want to thank the Flemish Fund for Scientific Research (FWO) for financial support in the FWO project G.0091.07. Also the financial support by the European Science Foundation (ESF) for the ORGANISOLAR Network Program ‘New Generation of Organic based Photovoltaic Devices' and for the EUROCORES (ESF) Program SONS (CRP SOHYDs) that is also supported by funds from the FWO (G.068506), the UHasselt (Research Fund) and the EC Sixth Framework Program under Contract ERAS-CT-2003-980409 is acknowledged.Notes and References
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- D. Braun and A. J. Heeger, Appl. Phys. Lett., 1991, 58, 1982 CrossRef CAS.
- P. L. Burn, A. B. Holmes, A. Kraft, D. D. C. Bradley, A. R. Brown, R. H. Friend and R. W. Gymer, Nature, 1992, 356, 47 CrossRef CAS.
- D. D. C. Bradley, Synth. Met., 1993, 54, 401 CrossRef CAS.
- S. Roth and F. Wudl, J. Am. Chem. Soc., 1996, 118, 10678 CrossRef.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741 CrossRef CAS.
- G. Horowitz, Adv. Mater., 1998, 10, 365 CrossRef CAS.
- A. G. MacDiarmid, W. J. Zhang, J. Feng, F. Huang and B. R. Hsieh, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1998, 39, 80 CAS.
- P. S. Heeger and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12219 CrossRef CAS.
- M. Gerard, A. Chaubey and B. D. Malhorta, Biosens. Bioelectron., 2002, 17, 345 CrossRef CAS.
- J. C. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15 CrossRef CAS.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924 CAS.
- H. Hoppe, M. Niggeman, C. Winder, J. Kraut, R. Heisgen, A. Hinsh, D. Meissner and N. S. Sariciftci, Adv. Funct. Mater., 2004, 14, 1005 CrossRef CAS.
- M. Besbes, G. Trippé, E. Levillain, M. Mazari, F. Le Derf, I. F. Perepichka, A. Derdour, A. Gorgues, M. Sallé and J. Roncali, Adv. Mater., 2001, 13, 1249 CrossRef CAS.
- I. Van Severen, F. Motmans, L. Lutsen, T. J. Cleij and D. Vanderzande, Polymer, 2005, 46, 5466 CrossRef CAS.
- M. Lanzi, P. Costa-Bizzarri, L. Paganin and G. Cesari, React. Funct. Polym., 2007, 67, 329 CrossRef CAS.
- H.-B. Bu, G. Goetz, E. Reinold, A. Vogt, S. Schmid, R. Blanco, J. L. Segura and P. Bauerle, Chem. Commun., 2008, 1320 RSC.
- T. L. Benanti, A. Kalaydjian and D. Venkataraman, Macromolecules, 2008, 41, 8312 CrossRef CAS.
- A. J. J. M. van Breemen, D. J. M. Vanderzande, P. J. Adriaensens and J. M. J. V. Gelan, J. Org. Chem., 1999, 64, 3106 CrossRef CAS.
- A. J. J. M. van Breemen, A. C. J. Issaris, M. M. de Kok, M. J. A. N. Van Der Borght, P. J. Adriaensens, J. M. J. V. Gelan and D. J. M. Vanderzande, Macromolecules, 1999, 32, 5728 CrossRef CAS.
- L. J. Lutsen, A. J. van Breemen, W. Kreuder, D. J. Vanderzande and J. M. J. V. Gelan, Helv. Chim. Acta, 2000, 83, 3113 CrossRef CAS.
- H. Roex, P. Adriaensens, D. Vanderzande and J. Gelan, Macromolecules, 2003, 36, 5613 CrossRef CAS.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686 CrossRef.
- M. Bjerring, J. S. Nielsen, N. C. Nielsen and F. C. Krebs, Macromolecules, 2007, 40, 6012 CrossRef CAS.
- S. A. Gevorgyan and F. C. Krebs, Chem. Mater., 2008, 20, 4386 CrossRef CAS.
- M. H. Petersen, S. A. Gevorgyan and F. C. Krebs, Macromolecules, 2008, 41, 8986 CrossRef CAS.
- A. J. Mozer, P. Denk, M. C. Scharber, H. Neugebauer, N. S. Sariciftci, P. Wagner, L. Lutsen and D. Vanderzande, J. Phys. Chem. B, 2004, 108, 5235 CrossRef CAS.
- E. Kesters, D. Vanderzande, L. Lutsen, H. Penxten and R. Carleer, Macromolecules, 2005, 38, 1141 CrossRef CAS.
- H. Sun, Z. Liu, Y. Hu, L. Wang, D. Ma, X. Jing and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2124 CrossRef CAS.
- C. Yang, J. Hou, B. Zhang, S. Zhang, C. He, H. Fang, Y. Ding, J. Ye and Y. Li, Macromol. Chem. Phys., 2005, 206, 1311 CrossRef CAS.
- C. Lee, Y. Kang, S. Hyun Jung, J.-S. Kim and J. Lee, Opt. Mater., 2003, 21, 337 CrossRef CAS.
- H. M. Barentsen, M. van Dijk, P. Kimkes, H. Zuilhof and E. J. R. Sudhölter, Macromolecules, 1999, 32, 1753 CrossRef CAS.
- H. J. Jung, Y. J. Park, S. H. Choi, J.-M. Hong, J. Huh, J. H. Cho, J. H. Kim and C. Park, Langmuir, 2007, 23, 2184 CrossRef CAS.
- G. Qu, F. Jiang, S. Zhang and S. Usuda, Mater. Lett., 2007, 61, 3421 CrossRef CAS.
- D. Xu, Z. Deng, X. Li, Z. Chen and C. Liang, Appl. Surf. Sci., 2007, 253, 3378 CrossRef CAS.
- J. Morgado, R. H. Friend and F. Cacialli, Synth. Met., 2000, 114, 189 CrossRef CAS.
- B.-L. Li, Z.-T. Liu, Y.-M. He, J. Pan and Q.-H. Fan, Polymer, 2008, 49, 1527 CrossRef CAS.
- Z. Li, Q. Zeng, G. Yu, Z. Li, C. Ye, Y. Liu and J. Qin, Macromol. Rapid Commun., 2008, 29, 136 CrossRef CAS.
- S. Campidelli, B. Ballesteros, A. Filoramo, D. Diaz Diaz, G. de la Torre, T. Torres, G. M. Aminur Rahman, C. Ehli, D. Kiessling, F. Werner, D. M. Guldi, C. Cioffi, M. Prato and J.-P. Bourgoin, J. Am. Chem. Soc., 2008, 130, 11503 CrossRef CAS.
- J. Feng, Q. Zhang, W. Li, Y. Li, M. Yang and Y. Cao, J. Appl. Polym. Sci., 2008, 109, 2283 CrossRef CAS.
- D. Quémener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155 CrossRef CAS.
- N. Luo, J. B. Hutchison, K. S. Anseth and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1885 CrossRef CAS.
- K. Nagesh and S. Ramakrishnan, Synth. Met., 2005, 155, 320 CrossRef CAS.
- N. Pérez-Moral and A. G. Mayes, Macromol. Rapid Commun., 2007, 28, 2170 CrossRef CAS.
- B. Sellergren, B. Rückert and A. J. Hall, Adv. Mater., 2002, 14, 1204 CrossRef CAS.
- N. A. Peppas and J. H. Ward, Adv. Drug Delivery Rev., 2004, 56, 1587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the functional monomer 5 as well as intermediates 1–4 and model compounds 9–17. See DOI: 10.1039/c0py00086h |
| This journal is © The Royal Society of Chemistry 2010 |