Synthesis, characterization and in vitro degradation of 3D-microstructured poly(ε-caprolactone) resins
Stefan
Theiler
a,
Michael
Teske
b,
Helmut
Keul
*a,
Katrin
Sternberg
*b and
Martin
Möller
*a
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen and DWI an der RWTH Aachen e.V., Pauwelsstr. 8, 52062, Aachen, Germany. E-mail: keul@dwi.rwth-aachen.de; moeller@dwi.rwth-aachen.de
bInstitute for Biomedical Engineering, University of Rostock, Friedrich-Barnewitz-Straße 4, 18119, Rostock, Germany. E-mail: katrin.sternberg@uni-rostock.de
First published on 9th June 2010
Abstract
Poly(ε-caprolactone)s of controlled molecular weight and low molecular weight distribution were prepared via anionic ring-opening polymerization using a tetra-functional initiator. The prerequisite for crosslinking was achieved by end-capping of the arms with acrylate groups. Novel biodegradable polyester resins were prepared by crosslinking of the functional polyesters via Michael addition using amino-telechelic poly(tetrahydrofuran). Three-dimensional microstructuring via replica molding shows the potential of this material as substrate for biomedical devices. Thermal and mechanical properties were investigated to characterize the polyester resins, accelerated in vitro degradation studies were carried out in a Sørensen buffer at pH 7.4 and 60 °C for up to 77 days. At different time intervals, the mass loss of the resins and the pH values of the buffer were determined, degradation products were investigated by means of NMR, SEC and ESI-MS and morphology of the degraded resins was checked via scanning electron microscopy. Compared to linear poly(ε-caprolactone) the degradation rate of all resins is higher, showing a mass loss of 50% within 77 days.
Introduction
In past decades biodegradable polymers have received increasing interest for use in biomedical applications. Aliphatic polyesters derived from cyclic monomers such as lactones or lactides are the most interesting biodegradable polymers and therefore several review papers deal with this subject.1–3 Due to their good mechanical properties, their biodegradability and biocompatibility,1,4,5 these polymers are widely used for biomedical applications such as surgical sutures,6 drug delivery systems7,8 and tissue engineering scaffolds.9Poly(ε-caprolactone) (PCL) is one of the most promising synthetic polyesters that can degrade in an aqueous medium under acidic or alkaline catalysis, respectively under enzymatic catalysis. The synthetic approach to PCL is ring-opening polymerization of ε-caprolactone using different nucleophilic initiators10,11 or enzymes such as lipases in aprotic media as catalysts.12 PCL provides a wide processing range starting from 100 °C which is 35 °C above the melting temperature of PCL (Tm(PCL) ≈ 65 °C) up to 200 °C which is well below the decomposition temperature of 350 °C. Furthermore its low glass transition temperature (Tg(PCL) ≈ −60 °C) provides PCL a rubbery consistency at room temperature.
Both enzymatic and chemical catalyzed hydrolytic degradation of PCL have been extensively investigated, showing that several lipases accelerate the degradation significantly,13–15 while hydrolytic degradation usually needs several years.16 Next to the mechanism of the hydrolytic degradation the erosion profiles of the polymers are of crucial importance for the successful implementation of these polymers in biomedical applications.17,18 The main steps involved in the erosion process are: (i) the diffusion of water into the bulk polymer, which is strongly affected by its porosity and might be accompanied by swelling of the material, (ii) the polymer degradation leading to the formation of cyclic oligomers and monomers which are water soluble at low concentration, and (iii) the weight loss via diffusion of degradation products from the bulk material. Concerning the erosion process, two types are distinguished, either heterogeneous surface or homogeneous bulk erosion.18 Crucial parameters are the diffusivity of water inside the matrix, the degradation rate of the functional groups of the back bone and the matrix dimensions respectively its porosity. Surface erosion occurs when the diffusion of water into the polymer is slower than the degradation of the polymer. In this case, only the region close to the surface of polymer matrix is affected by hydrolysis. In course of this process, the hydrolysis front advances towards the material core, since the surface is eroded and removed continuously. This type of erosion behavior is generally shown by poly(ortho ester)s, poly(anhydride)s and some poly(carbonate)s.19,20
In the case of bulk erosion, water penetrates throughout the entire polymer matrix before hydrolysis occurs. Consequently, chain cleavage happens uniformly throughout the entire material. The mechanical properties change from the very beginning, in parallel with mass loss and the decrease of the molecular weight. Typical polymers following this erosion mechanism are poly(lactic acid) and poly(ε-caprolactone).
The degradation rate is influenced by various factors. It is generally accepted that hydrolysis of ester groups initially occurs in the amorphous domains of the polymer. The degradation rate increases when the crystallinity of the polymer decreases. An increasing molecular weight results in a lower degradation rate. Furthermore the hydrophilicity of the material, crosslinking density, porosity and surface chemistry may influence the degradation rate.21–23 This high number of parameters influencing the degradation rate of polymers results in a wide range of possibilities to tailor this rate. For demonstration some examples are given: (i) incorporation of a hydrophilic monomer into a hydrophobic polyester increases the rate of degradation; this was achieved by copolymerization of 1,5-dioxepan-2-one with ε-caprolactone. (ii) By using a hydrophilic macroinitiator for the polymerization of ε-caprolactone, e.g. a hydroxy-telechelic poly(ethylene oxide), amphiphilic block copolymers with enhanced degradation rate are obtained.24–26 (iii) Changing the microstructure and architecture of PCLs by preparation of block or graft copolymers, star-shaped polymers, porous scaffolds and crosslinked networks, leads to a decrease in crystallinity and an increase of the degradation rate.27–29 (iv) Modification of the end groups, e.g. by attaching carboxylic acid groups at the chain end enhances moisture uptake and hence accelerates the degradation.30–32
This latter possibility is limited for linear polymers to two functional groups (the chain ends). By changing the architecture of the polymer from linear to star-shaped or brush like molecules the number of functional end groups can be increased. Compared to linear PCLs, star-shaped PCLs have a lower crystallinity and lower melt viscosity.33 Star-shaped PCLs were prepared starting with pentaerythritol, dipentaerythritol or star-shaped poly(ethylene glycol) and using tin or zinc based catalyst.34–38 By end functionalization of these star-shaped polymers with acrylate,39 methacrylate,40,41 maleate or itaconate42 end groups the prerequisites for the preparation of crosslinked poly(ε-caprolactones) was accomplished. Photochemical or thermal radical crosslinking reactions were performed and the hydrolytic degradation of these materials was studied. In accordance with the lower crystallinity induced by the crosslinking a higher degradation rate was obtained.
Furthermore, crosslinked polyesters were obtained from DL-lactide oligomers end capped with triethoxysilane groups via hydrolytic polycondensation.43
In this paper we present a simple synthetic route towards functional star-shaped poly(ε-caprolactone)s end-capped with acrylate groups. The star-shaped PCLs of different molecular weights were crosslinked via Michael addition with amino-telechelic poly(tetrahydrofuran), as a model diamine, resulting in novel polyester/polyether composite materials. Using the replica molding technique, 3D-microstructured degradable coatings with uniform topography were prepared. Furthermore the resins were characterized with regard to thermal and mechanical properties, and hydrolytic degradation behavior, in order to see the influence of the resin composition on these properties.
Experimental section
Materials
ε-Caprolactone (CL, ≥99%, Fluka) was stirred over CaH2 for 24 h, condensed onto molecular sieves (4 Å), and kept in a Schlenk flask under argon. Di(trimethylolpropane) (diTMP, 97%, Aldrich) was purified by sublimation and kept under argon. Zinc(II) 2-ethylhexanoate (Zn(oct)2, 97%, ABCR), acryloyl chloride (96%, Merck) and poly(tetrahydrofuran) bis(3-aminopropyl) terminated (Mn = 1100 g mol−1) (Aldrich) were used as received.All polymerizations were carried out in an inert gas atmosphere. Argon (Linde) was passed over molecular sieves (4 Å) and finely distributed potassium on aluminium oxide.
Measurements
1H and 13C NMR spectra were recorded on a Bruker AV 400 FT-NMR spectrometer at 400 MHz and 133 MHz, respectively. Deuterated chloroform (CDCl3) was used as the solvent, and tetramethylsilane (TMS) served as internal standard.Size exclusion chromatography (SEC) analyses were carried out using THF (with addition of 250 mg L−1 2,6-di-tert-butyl-4-methylphenol) as eluting solvents. A high pressure liquid chromatography pump (ERC HPLC model 64200) and a refractive index detector (Jasco RI-2031) were used at 30 °C with a flow rate of 1.0 mL min−1. Four columns with MZ-DVB gel were applied. The length of each column was 300 mm and the diameter 8 mm, the diameter of the gel particles was 5 μm, and the nominal pore widths were 50, 102, 103 and 104 Å. Calibration was achieved using narrow distributed poly(methyl methacrylate) (PMMA) standards.
Differential scanning calorimetry (DSC) was performed on a Netzsch DSC 204. The measurements were performed in air with a heating rate of 10 K min−1. Calibration was achieved using indium and cyclohexane.
Raman Spectroscopy was carried out on a Bruker FT-Raman-Spectrometer RFS 100/S with a Nd:YAG-laser. The power of the laser was 200 mV at 1064 nm with a spectral resolution of 4 cm−1. 500 scans were performed for each spectrum.
Synthesis
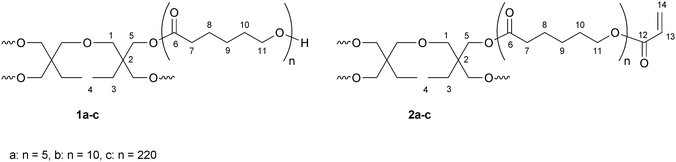 | ||
| Scheme 1 Structure of sPCL 1a–c and sPCL-A 2a–c, with numbers of NMR assignment. | ||
1H NMR (CDCl3): δ (ppm) = 0.80–0.92 (m, H4); 1.32–1.46 (m, H3, H9); 1.58–1.72 (m, H8, H10); 2.31 (tr, H7); 3.22–3.42 (m, H1); 3.60–3.70 (tr, H11E); 3.96–4.14 (m, H5, H11).
13C NMR (CDCl3): δ (ppm) = 24.6 (C8); 24.7 (C8E); 25.3 (C9E); 25.5 (C9); 28.3 (C10); 32.3 (C10E); 34.1 (C7); 34.2 (C7E); 62.6 (C11E); 64.1 (C11); 173.5 (C6); 173.7 (C6E) (the peaks of the initiator are not distinguishable from the noise of the baseline).
From the 1H NMR spectra a degree of polymerization of DPn = 20, respectively 37 or 854 was determined (Mn,NMR = 2500 (1a), and 4800 (1b) and 100700 (1c)).
1H NMR (CDCl3): δ (ppm) = 0.80–0.92 (m, H4); 1.32–1.48 (m, H3, H9); 1.58–1.72 (m, H8, H10); 2.31 (tr, H7); 3.22–3.36 (m, H1); 3.96–4.10 (m, H5, H11); 4.10–4.20 (tr, H11E) 5.82 (d, H14); 6.11 (dd, H13); 6.40 (d, H14).
13C NMR (CDCl3): δ (ppm) = 24.6 (C8); 25.5 (C9); 28.3 (C10); 34.1 (C7); 64.1 (C11); 64.3 (C11E); 128.5 (C13); 130.6 (C14); 166.2 (C12); 173.7 (C6) (the peaks of the initiator are not distinguishable from the noise of the baseline).
From the 1H NMR spectra a degree of functionalization of the hydroxy groups of 100% was determined (Mn,NMR = 2500 (2a), 4900 (2b) and 133700 (2c)).
PDMS master fabrication
The poly(dimethylsiloxane) (PDMS) prepolymer and curing agent (Slygard 184, Dow Corning) were mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (v/v), degassed under vacuum, poured onto a silicon master, and cured at 80 °C for 2 h. The PDMS replica was then carefully peeled off. In this way PDMS masters with inverse geometry (holes and ridges) were obtained.
:1 ratio (v/v), degassed under vacuum, poured onto a silicon master, and cured at 80 °C for 2 h. The PDMS replica was then carefully peeled off. In this way PDMS masters with inverse geometry (holes and ridges) were obtained.
Replica molding
Polyesters were microstructured by thermal UV replica molding using elastic PDMS or rigid Teflon masters. Solutions of the star-shaped acrylate functional poly(ε-caprolactone) (30–50 wt% in dichloromethane) and amino-telechelic poly(tetrahydrofuran) in dichloromethane were dispensed on the master, heated, and carefully peeled off.Degradation
Mechanical tests
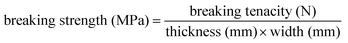 | (1) |
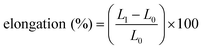 | (2) |
Results and discussion
Crosslinked biodegradable polymers are important scaffolds used in biomedical application. Recently we reported on the preparation of biodegradable polyester resins via photocrosslinking of functional polyesters. Linear poly(caprolactone)s with randomly distributed γ-acryloyloxy-ε-caprolactone and γ-methacryloyloxy-ε-caprolactone repeating units44 or star-shaped PCLs end-capped with methacrylate groups were used as soluble precursors. To increase the number of functional end groups bottle brushes were prepared having a poly(glycidol) backbone and poly(caprolactone) or poly(lactide) side chains partially end-capped with acryloyl groups.45 Crosslinking of the polyesters decreases their crystallinity, resulting in materials with enhanced degradation rate and improved mechanical properties, for example materials with reduced brittleness.In this paper, a different crosslinking reaction was used for the preparation of polyester resins: Michael addition of diamines to acrylate terminated PCL star polymers. The synthetic concept comprises three steps (Scheme 2). (i) Ring-opening polymerization of CL using a tetrafunctional initiator, (ii) end-capping of the PCL star arms and (iii) crosslinking via Michael addition using an amino telechelic poly(tetrahydrofuran) as model diamine. The chemical nature, the molecular weight and the mass fraction of different diamines used will influence the resin properties. Furthermore a partial degradation, obtained due to crosslinking of a degradable with a non-degradable polymer, might result in minor loss of the mechanical stability of test specimens during degradation. However, although the polyether is non-degradable it can be eliminated from the organism due to its low molecular weight.
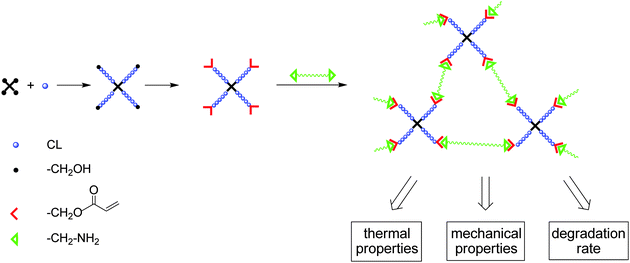 | ||
| Scheme 2 Synthetic concept for the preparation of polyester resins. | ||
Synthesis
Star-shaped PCLs were synthesized using di(trimethylolpropane) as tetrafunctional initiator and zinc 2-ethylhexanoate as catalyst. The ratio of CL repeating units to the hydroxymethyl end groups can easily be adjusted by the monomer/initiator ratio in feed. In the second step, the polymer was functionalized via Schotten–Baumann reaction with acryloyl chloride resulting in functional star-shaped PCL with acrylate end groups (sPCL-A) (Scheme 3).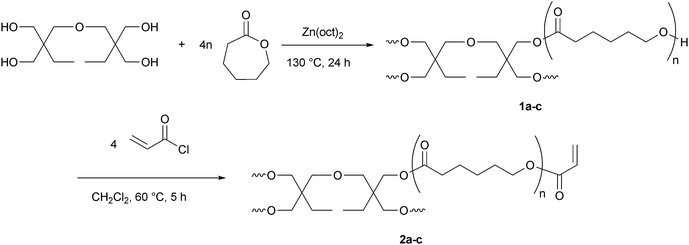 | ||
| Scheme 3 Synthetic pathway to star-shaped functional poly(ε-caprolactone) (sPCL-A). | ||
The 1H NMR spectrum of sPCL 1a (Fig. 1a) shows the expected signals: the CL repeating units (signals 7, 8, 9, 10 and 11), the hydroxymethyl end group (signal 11E) and the signals of the initiator. The number of CL repeating units in the star-shaped PCL is determined by comparison of a signal of the initiator (e.g. H4) with a signal of the CL repeating units (e.g. H7), the total amount of CL repeating units (DPn) is determined as 20.
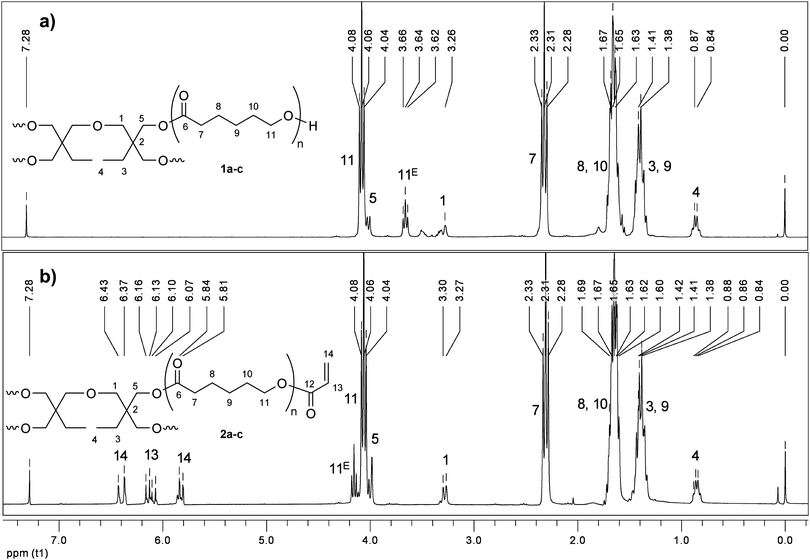 | ||
| Fig. 1 1H NMR spectra of star-shaped PCL 1a and sPCL-A 2a. | ||
The polymers were obtained in yields > 97%. Typical molecular weight distributions obtained via SEC analysis were 1.3–1.4 (Table 1).
| [M]/[I]a | Yield (%) | M n,theory | M n,NMR | M n,SEC | M w/Mnd | DPne | |
|---|---|---|---|---|---|---|---|
| a Monomer to initiator ratio in feed. b Calculated molecular weight at full conversion. c Number average molecular weight (Mn) determined by NMR. d M n and molecular weight distribution (Mw/Mn) determined by SEC (PS standards). e Total number of repeating units. | |||||||
| 1a | 20 | 100 | 2500 | 2500 | 4900 | 1.31 | 20 |
| 1b | 40 | 97 | 4800 | 4400 | 9500 | 1.41 | 37 |
| 1c | 880 | 97 | 100700 | 97700 | 141200 | 1.45 | 854 |
Successful functionalization with acryloyl chloride can be proven by the appearance of the signals of the functional groups (H13, H14). Furthermore the signal of the CL end group (H11E) is shifted to lower field. Since the whole signal shifted (no residual signal observed), the functionalization is quantitative, resulting in a star-shaped PCL bearing acrylate end groups (sPCL-A 2a).
Table 2 summarizes the results obtained upon functionalization, resulting in polymers with a degree of functionalization (DFn) of 100% and similar molecular weights and molecular weight distributions as in the starting polymers 1a–c.
| Yield (%) | DFa (%) | M n,NMR | M n,SEC | M w/Mnc | |
|---|---|---|---|---|---|
| a Degree of functionalization determined by NMR. b Number average molecular weight (Mn) determined by NMR. c M n and molecular weight distribution (Mw/Mn) determined by SEC (PS standards). | |||||
| 2a | 100 | 100 | 2700 | 4300 | 1.42 |
| 2b | 100 | 100 | 4600 | 9000 | 1.40 |
| 2c | 100 | 100 | 97900 | 133700 | 1.58 |
Crosslinking reaction
Novel biodegradable polyester resins were prepared by thermal crosslinking of the acrylate functionalized star-shaped poly(caprolactone)s. Crosslinking of the acrylate groups with amino-telechelic poly(tetrahydrofuran) via Michael addition was carried out on polymer films from a mixture of both functional polymers. Quantitative conversion is observed yielding an insoluble resin (RCL-A), proving successful crosslinking. Scheme 4 exemplifies the chemistry of the crosslinking reaction.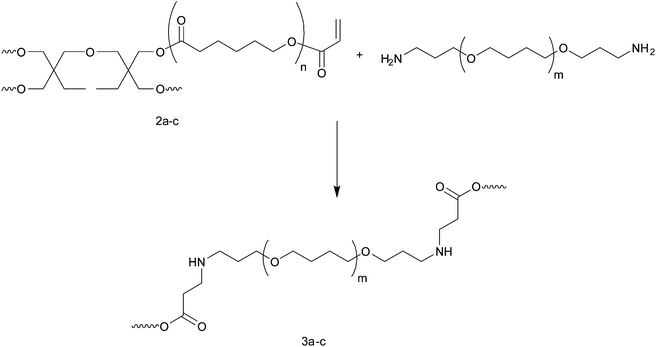
Replica molding
Replica molding consists of the replication of microstructured substrates in polymeric materials, presenting the inverse geometry of the one aimed at. This micropatterning technique offers several advantages: it is a relatively inexpensive technique fast and easy to execute, consisting of only three main steps: preparing of the polymer/diamine solution, thermal crosslinking, and release of the replica (Scheme 5). | ||
| Scheme 5 A schematic representation of the thermal replica molding process. | ||
The only expensive step of the whole process is the fabrication of the master as it requires cleanroom facilities. However, it can be used many times and with the so-called secondary mold approach, where a polymeric replica of the master is used as the new mold to be replicated, the repetitive use of the (expensive) master is limited. Advantage of this approach was taken by using PDMS molds as secondary masters.
Large area patterns were made from a solution of sPCL-A (2a–c) in dichloromethane (20 wt%) using amino-telechelic poly(tetrahydrofuran) as crosslinking agent (0.5 eq.), heating to 55 °C for 15 h, and carefully peeling off. Replicas with pillars (10 μm diameter, 9 μm height and 20 μm interdistance) and ridges (5 μm grooves, 15 μm ridges) were prepared. On the same masters, also replica molding by solvent evaporation without any chemical crosslinking was performed, SEM images are shown in Fig. 2. Replicas from both low and high molecular weight polymers were fabricated. In the case of the low molecular weight, only crosslinked films could be released from the PDMS masters as self-supporting micropatterned substrates. In the case of the high molecular weight polymer, the fidelity of replication was higher (the surface was smoother) and both crosslinked and uncrosslinked films could be released from the masters and handled without problems.
 | ||
| Fig. 2 Large area patterns of the resins/polyester. SEM images of crosslinked RCL-A 3a–c (a–c) and uncrosslinked sPCL-A 2c (d). | ||
According to the results obtained with linear functional polyesters,44 the quality of the replica increases with increasing crosslinking density, or in other words, with decreasing the ratio of CL repeating units to crosslinkable functional groups in the polymer, as shown in Fig. 2. The resins prepared by crosslinking of polymers with a low ratio of CL repeating units to acrylate groups were transparent and tough but still elastic (a, b). This is attributed to the amorphous state of the resins, which results from the high density of crosslinking. Using high molecular weight functional polymers, opaque and brittle resins with less defined features were obtained (c, d). Both the resin having a relative low crosslinking density (c) and the uncrosslinked polymer (d), are semicrystalline. The crystallinity of the samples is responsible for both the opacity and the brittleness. In case of crystalline materials with a low concentration of crosslinkable groups, crosslinking does not have an influence in this microstructuring process and the material's properties. The long PCL chains control the properties of both, the star-shaped polyester and the resin, since crosslinking density in the resin is too low.
Thermal analysis
The thermal properties – glass transition and melting temperature – of the functional polyesters and the corresponding resins were determined and compared using DSC. The influence of the ratio CL repeating units to acrylate groups in the polyesters and the influence of the ratio CL repeating units to THF repeating units for the resins were investigated. Exemplarily, Fig. 3 demonstrates the DSC (second heating and cooling curve) of sPCL-A 2a and the corresponding resin RCL-A 3a. The major difference between the two DSC traces is that sPCL-A 2a shows two melting transitions characteristic for oligo caprolactone while RCL-A 3a presents a pronounced glass transition temperature at −52 °C and a vague melting point.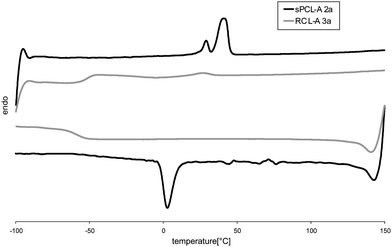 | ||
| Fig. 3 DSC second heating and cooling curves of sPCL-A 2a and the corresponding resin RCL-A 3a. | ||
In the cooling scan, crystallization is found for sPCL-A 2a at ∼0 °C and for RCL-A 3a again only the glass transition is observed.
All polyesters 2a–c are semicrystalline polymers (Table 3). Glass transition temperatures are unincisive, they appear at −61 °C to −62 °C. Melting temperatures in the region of 29/41 °C (sPCL-A 2a) to 58 °C (sPCL-A 2c) were found, which are typical values for oligo- and poly(ε-caprolactone)s. Increasing the DPn of the polymers, the (longer) chains are able to form larger crystallites resulting in higher melting temperatures and a higher melting enthalpy (48 J g−1 (sPCL-A 2a) to 79 J g−1 (sPCL-A 2c)). The resins show clear glass transitions and melting transitions of low enthalpy. This indicates that crosslinking of the polymers prevents the formation of crystallites which results in a strong increase of the amorphous character of the material. RCL-A 3a is amorphous and has only residual crystallinity (ΔHm = 2 J g−1), while RCL-A 3b shows a slight decrease of the melting enthalpy (ΔHm = 43 J g−1) and 3c possesses almost unchanged crystallinity (ΔHm = 70 J g−1). Resins 3a and 3b show a higher glass transition temperature than their precursors reflecting the lower mobility of the short PCL chains in the resin. Crosslinking density of 3c is too low, to achieve any significant change in thermal properties after crosslinking.
It is our aim to prepare biodegradable materials with tunable degradation rate and suitable/predefined mechanical properties. However, the degradation rate and the mechanical stability have to be adjusted on each other. Crystallinity of the material influences both, degradation rate and mechanical properties, but contrarily: increasing crystallinity decreases degradation rate and enhances the mechanical properties.
Biodegradable polymers are mostly aliphatic polyesters. These polymers are semicrystalline with a low glass transition temperature (well below ambient temperature). Their mechanical stability is mainly defined by the crystalline domains of the materials. In order to increase the degradation rate of aliphatic polyesters crystallinity has to be reduced; in order to compensate the loss of mechanical stability a crosslinked polyester was chosen.
In the following, the compromise between enhanced degradation rate and loss in mechanical stability on the crosslinked poly(caprolactone)s is described.
Test specimen preparation for degradation and mechanical tests
To prepare test specimens, sPCL-A samples were crosslinked with amino-telechelic poly(tetrahydrofuran) by replica molding using a Teflon master. For comparison reasons, linear PCL test specimens were used. For degradation experiments and for suture retention strength and burst pressure measurements rectangular blocks were used (see Experimental section), while for tensile strength measurements shouldered test bars were prepared. The crosslinking density was determined by Raman spectroscopy, comparing the concentration of C,C double bonds in the sPCL-A samples and in the corresponding resins.The composition of the resins and the degree of crosslinking determined by Raman spectroscopy are summarized in Table 4. In both cases the acrylate groups were completely converted. Although this observation does not indicate that all consumed C,C double bonds contributed to crosslinking it can be assumed that crosslinked networks were formed, since the resins are insoluble in organic solvents such as chloroform.
Degradation
The degradation of poly(ε-caprolactone) is widely investigated since this material is used for many biomedical applications. Biocompatibility, good permeability for many therapeutic drugs in combination with a low degradation rate make it suitable as matrix for drug delivery systems, which remain active for more than one year.4 It has been shown, that degradation preferentially occurs in the amorphous domains since the water cannot penetrate the crystalline zones.46 Increasing the amorphous domains of a PCL based material should increase the degradation rate and thus make it suitable for new applications.As already shown before, crosslinking of sPCL-A with diamines results in resins with a lower crystallinity than linear PCL. However, the difference in crystallinity is rather small for the high molecular weight samples (3cvs.2c). Therefore in the following the hydrolytic degradation and erosion behavior of only resins 3a and 3b was studied, with respect to the mass loss, the change of pH and morphology. A relationship between the crosslinking density of the resins and its degradation rate was determined. The materials were compared to linear PCL (Mn = 80000) as reference since this material is usually used in biomedical applications.
The degradation rates of RCL-A 3a and 3b (degradation performed in Sørensen buffer at pH 7.4 and 60 °C according to ISO-Norm 15814) were faster than that of pure PCL. A total mass loss of 48% (RCL-A 3a) and 55% (RCL-A 3b) were detected. Pure PCL showed a mass loss of only 5% after 77 days of degradation (Fig. 4). The curve progressions of the mass loss were almost linear for both resins, indicating a continuous degradation behavior.16,18
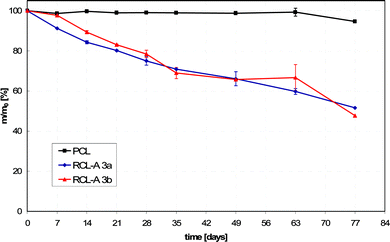 | ||
| Fig. 4 Degradation-induced mass loss of RCL-A 3a and 3b in comparison to PCL under accelerated conditions at 60 °C in Sørensen buffer (0.1 M, pH 7.4). | ||
Degradation of PCL containing resins occurs via hydrolysis of the ester bonds, resulting in the formation of 6-hydroxyhexanoic acid. During degradation it is expected that the pH of the reaction medium decreases due to increase of the concentration of carboxylic acid groups. The pH change during the degradation of RCL 3a and 3b, in which the medium was not changed throughout the degradation time, reveals a linear decrease for both samples (Fig. 5). These curve progressions confirm the assumption of a continuous bulk degradation, in which acidic byproducts are removed from the polymer matrix. These findings correspond to established results of the accelerated in vitro degradation of PCL.47
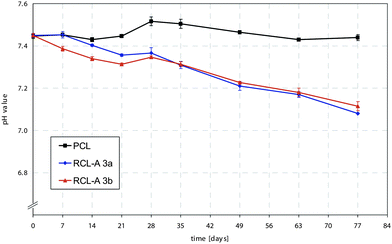 | ||
| Fig. 5 Degradation-induced changes of pH value of RCL-A 3a and 3b in comparison to PCL under accelerated conditions at 60 °C in Sørensen buffer (0.1 M, pH 7.4). | ||
Analysis of the soluble degradation products via NMR, SEC and ESI-MS reveals the presence of poly(THF) crosslinking agent (Mn1 = 1100 g mol−1), of partly degraded sPCL (Mn2 = 6500 g mol−1) and of 6-hydroxyhexanoic acid.
The degradation behavior of RCL-A 3a and RCL-A 3b were further investigated via macro photography and SEM (Fig. 6 and 7). The morphological observations show the similarity in degradation of both polymeric materials. The sample structure of RCL-A 3a was already lost within the first week. RCL-A 3b lost its geometry within the first 14 days of the accelerated degradation. The microscopic investigations confirmed these results (Fig. 6 (A`–C`) and 7 (A`–C`)). The geometry loss of the polymers after the first week of degradation leads to a mainly smooth surface. As it is shown in Fig. 6 A`–B` and 7 A`–B`, the roughness of the starting material gets almost lost. Within the ongoing degradation, the polymer roughness increased again, this is most probably caused by removal of fragments (Fig. 6 C` and 7 C`). Further degradation leads to the formation of a white powdery residue was obtained. Micro cracks or holes were hardly feasible during the degradation process. Due to the geometry loss, the surface of the samples is increased and the diffusion of water into the inner parts of the resin is easier. Therefore, most probably a continuous degradation is taking place, which result in a linear pH value change and mass loss.
 | ||
| Fig. 6 Surface morphology of RCL-A 3a after (a) 0, (b) 7, (c) 77 days accelerated in vitro degradation investigated by macro photography (A, B, C) and SEM (A′, B′, C′). | ||
 | ||
| Fig. 7 Surface morphology of RCL-A 3b after (a) 0, (b) 7, (c) 77 days accelerated in vitro degradation investigated by macro photography (A, B, C) and SEM (A′, B′, C′). | ||
Mechanical properties
Synthetic polymeric materials for blood vessels ideally have to show the same mechanical properties as natural ones. Since in most cases the complex property profile of natural constructs is out of reach, synthetic materials should at least aim for a certain level of these properties. Standard tests such as tensile strength, suture retention and burst pressure tests were performed on the resins 3a and 3b with regard to a potential application as material for vascular prostheses. Since the waxy polymers 2a and 2b obviously do not exhibit mechanical stability, these materials were not tested. For comparison reason linear PCL (Mn = 80000) was investigated as reference since it is usually used in biomedical applications.The tensile properties of the resins (Table 5) are significantly different from the ones of the high molecular weight PCL (Mn = 80000). RCL-A 3a and 3b have lower breaking strength (1.3 ± 0.1 MPa (3a) and 7.7 ± 0.8 MPa (3b)) and lower elongations at break (134 ± 11% (3a) and 622 ± 28% (3b)). E moduli of 1.2 ± 0.5 MPa (3a) 19.3 ± 1.6 MPa exhibit, that both resins are rubber-like materials, which is attributed to the fact, that the materials are in particular amorphous. Decreasing crosslinking density (increasing the length of PCL chains) leads to slightly stiffer materials. Elastic properties of the resins are in the range of natural devices. While 3a exhibits similar properties as fresh carotids (breaking strength: 2.0 ± 0.1 MPa, elongation at break: 158 ± 11%)48 and as human coronary arteries (E modulus: 1.0–2.0 MPa),493b even exhibits better mechanical stability.
| Tensile properties | Suture retention strength (MPa) | Burst pressure/bar | |||
|---|---|---|---|---|---|
| Breaking strength (MPa) | Elongation at break (%) | E modulus (MPa) | |||
| a Crosslinking conditions: 15 h at 55 °C. b Mn = 80000. c Ref. 48. d Ref. 50. e Ref. 51. f Ref. 49. | |||||
| RCL-A 3aa | 1.3 ± 0.1 | 134 ± 11 | 1.2 ± 0.5 | 1.1 ± 0.1 | 2.9 ± 0.3 |
| RCL-A 3ba | 7.7 ± 0.8 | 622 ± 28 | 19.3 ± 1.6 | 24.7 ± 3.9 | > 5.00 |
| PCLb | 23.3 ± 0.9 | 749 ± 23 | 253.9 ± 21.9 | 134.7 ± 12.9 | > 5.00 |
| Carotid artery | 2.0 ± 0.1c | 158 ± 11c | 140.6 ± 21.0d | 1.7 ± 0.2e | |
| Coronary artery | 1.0–2.0f | ||||
Suture retention strength is a crucial factor in the fabrication of materials for vascular prostheses as it directly relates to the graft implantation procedure. In this study, the resin RCL-A 3a had rather low suture retention strength of 1.1 ± 0.1 MPa. The resin RCL-A 3b exhibits much better suture retention strength (24.7 ± 3.9 MPa) due to the higher concentration of crystalline parts. Thus it can easily keep up with common electrospun PCL scaffolds52 or poly(lactide-co-glycolide)/polyurethane based vascular grafts.53 The suture retention strength of high molecular weight PCL is much higher (134.7 ± 12.9 MPa), which is rather in the region of the values of native carotid arteries (140.6 ± 21.0 MPa)50 or the values of native pig blood vessels.54
In order to determine whether the resins possessed adequate strength to endure physiological forces, burst pressure testing was performed to measure the maximum pressure that the material could endure before failure. As shown in Table 5, RCL-A 3a has a burst pressure of 2.9 ± 0.3 bar (2150 ± 250 mmHg). Increasing the amount of crystalline parts in the resin increase the burst pressure, since RCL-A 3b already has a burst pressure above 5 bar (4500 mmHg), when the measurement was limited by the experimental setup. This is a remarkable value, since native carotid arteries and native pig vessels, tested with the same setup, showed a burst pressure of 1.7 ± 0.2 bar (1275 ± 122 mmHg),51 and 3.4–4.1 bar (2550–3100 mmHg)54 respectively. PCL also did not burst, until the maximum applicable pressure was reached.
Conclusions
Star-shaped poly(ε-caprolactone)s of different molecular weights bearing acrylate end groups were prepared via anionic ring-opening polymerization of ε-caprolactone using a tetrafunctional initiator followed by end-capping with acryloyl chloride. The ratio of CL repeating units per functional end group can easily be adjusted by the monomer/initiator ratio. The acrylate end groups were used as substrates for crosslinking reactions via Michael addition. Using amino-telechelic poly(tetrahydrofuran) as crosslinking agent polyester/polyether resins were prepared. Using replica molding precise micropatterned surfaces with high resolution were obtained. Due to their degradation rate and their mechanical properties the polyester/polyether resins are promising candidates for the development of vascular prostheses.Acknowledgements
The authors thank Stefanos Diamantouros from Dept. of Applied Medical Engineering, Helmholtz Institute, RWTH Aachen for performing the mechanical tests.This research was carried out in the framework of the SFB Transregio 37 project “Mikro- und Nanosystheme in der Medizin - Rekonstruktion biologischer Funktionen” funded by the German Research Foundation.
References
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- A. Duda and S. Penczek, in Biopolymers, ed. A. Steinbüchel and Y. Doi, Wiley-VCH, Weinheim, 2002, vol. 3b, pp. 371–429 Search PubMed.
- S. Matsumura, Advances in Polymer Science, Vol. 194, Springer, Berlin/Heidelberg, 2006, pp. 95–132 Search PubMed.
- M. E. Gomes and R. L. Reis, Int. Mater. Rev., 2004, 49, 261–273 Search PubMed.
- M. E. Gomes and R. L. Reis, Int. Mater. Rev., 2004, 49, 274–285 Search PubMed.
- E. Behravesh, A. W. Yasko, P. S. Engel and A. G. Mikos, Clin. Orthop. Relat. Res., 1999, 367, S118–129 CrossRef.
- R. Langer, Science, 1990, 249, 1527–1533 CrossRef CAS.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- O. E. Teebken and A. Haverich, Eur. J. Vasc. Endovasc. Surg., 2002, 23, 475–485 CrossRef.
- T. Biela, A. Kowalski, J. Libiszowski, A. Duda and S. Penczek, Macromol. Symp., 2006, 240, 47–55 CrossRef CAS.
- H. Höcker and H. Keul, Adv. Mater., 1994, 6, 21–36 CrossRef.
- M. Hans, P. Gasteier, H. Keul and M. Möller, Macromolecules, 2006, 39, 3184–3193 CrossRef CAS.
- H. Fukuzaki, M. Yoshida, M. Asano, M. Kumakura, T. Mashimo, H. Yuasa, K. Imai and H. Yamanaka, Polymer, 1990, 31, 2006–2014 CrossRef CAS.
- M. Mochizuki, M. Hirano, Y. Kanmuri, K. Kudo and Y. Tokiwa, J. Appl. Polym. Sci., 1995, 55, 289–296 CrossRef CAS.
- Z. H. Gan, D. H. Yu, Z. Y. Zhong, Q. Z. Liang and X. B. Jing, Polymer, 1999, 40, 2859–2862 CrossRef CAS.
- A. Göpferich, Biomaterials, 1996, 17, 103–114 CrossRef CAS.
- M. Vert, E-Polymers, 2005, 008 Search PubMed.
- F. von Burkersroda, L. Schedl and A. Gopferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS.
- K. W. Leong, B. C. Brott and R. Langer, J. Biomed. Mater. Res., 1985, 19, 941–955 CrossRef CAS.
- T. Freier, C. Kunze, C. Nischan, S. Kramer, K. Sternberg, M. Sass, U. T. Hopt and K. P. Schmitz, Biomaterials, 2002, 23, 2649–2657 CAS.
- S. M. Li, H. Garreau and M. Vert, J. Mater. Sci.: Mater. Med., 1990, 1, 198–206 CrossRef CAS.
- M. Kallrot, U. Edlund and A. C. Albertsson, Biomacromolecules, 2007, 8, 2492–2496 CrossRef.
- M. Hakkarainen, A. Hoglund, K. Odelius and A. C. Albertsson, J. Am. Chem. Soc., 2007, 129, 6308–6312 CrossRef CAS.
- S. Karlsson, M. Hakkarainen and A. C. Albertsson, J. Chromatogr., A, 1994, 688, 251–259 CrossRef CAS.
- S. M. Li, P. Dobrzynski, J. Kasperczyk, M. Bero, C. Braud and M. Vert, Biomacromolecules, 2005, 6, 489–497 CrossRef CAS.
- S. S. Shah, K. J. Zhu and C. G. Pitt, J. Biomater. Sci., Polym. Ed., 1994, 5, 421–431 CAS.
- M. Hans, H. Keul and M. Möller, Biomacromolecules, 2008, 9, 2954–2962 CrossRef CAS.
- K. Odelius, A. Finne and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 596–605 CrossRef CAS.
- K. Odelius, P. Plikk and A. C. Albertsson, Biomacromolecules, 2005, 6, 2718–2725 CrossRef CAS.
- N. Andronova, R. K. Srivastava and A. C. Albertsson, Polymer, 2005, 46, 6746–6755 CrossRef CAS.
- K. R. Huffman and D. J. Casey, J. Polym. Sci., Part A: Polym. Chem., 1985, 23, 1939–1954 Search PubMed.
- J. S. Wiggins, M. K. Hassan, K. A. Mauritz and R. F. Storey, Polymer, 2006, 47, 1960–1969 CrossRef CAS.
- E. S. Kim, B. C. Kim and S. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 939–946 CrossRef CAS.
- K. R. Gorda and D. G. Peiffer, J. Appl. Polym. Sci., 1993, 50, 1977–1983 CrossRef CAS.
- M. A. R. Meier and U. S. Schubert, E-Polymers, 2005, 085 Search PubMed.
- J. L. Wang, L. Wang and C. M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5449–5457 CrossRef CAS.
- K. H. Kim, G. H. Cui, H. J. Lim, J. Huh, C. H. Ahn and W. H. Jo, Macromol. Chem. Phys., 2004, 205, 1684–1692 CrossRef CAS.
- M. X. Deng, X. S. Chen, L. H. Piao, X. F. Zhang, Z. L. Dai and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 950–959 CrossRef CAS.
- H. Kweon, M. K. Yoo, I. K. Park, T. H. Kim, H. C. Lee, H. S. Lee, J. S. Oh, T. Akaike and C. S. Cho, Biomaterials, 2003, 24, 801–808 CrossRef CAS.
- R. F. Storey, S. C. Warren, C. J. Allison, J. S. Wiggins and A. D. Puckett, Polymer, 1993, 34, 4365–4372 CrossRef CAS.
- A. O. Helminen, H. Korhonen and J. V. Seppälä, Macromol. Chem. Phys., 2002, 203, 2630–2639 CrossRef CAS.
- M. P. K. Turunen, H. Korhonen, J. Tuominen and J. V. Seppälä, Polym. Int., 2002, 51, 92–100 CrossRef CAS.
- A. Helminen, H. Korhonen and J. V. Seppälä, Polymer, 2001, 42, 3345–3353 CrossRef CAS.
- C. Vaida, P. Mela, H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6789–6800 CrossRef CAS.
- H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3209–3231 CrossRef CAS.
- C. Eldsater, B. Erlandsson, R. Renstad, A. C. Albertsson and S. Karlsson, Polymer, 2000, 41, 1297–1304 CrossRef CAS.
- C. X. F. Lam, M. M. Savalani, S. H. Teoh and D. W. Hutmacher, Biomed. Mater., 2008, 3, 034108 Search PubMed.
- A. Kurane, D. T. Simionescu and N. R. Vyavahare, Biomaterials, 2007, 28, 2830–2838 CrossRef CAS.
- I. Ozolanta, G. Tetere, B. Purinya and V. Kasyanov, Med. Eng. Phys., 1998, 20, 523–533 CrossRef CAS.
- S. W. Cho, S. H. Lim, I. K. Kim, Y. S. Hong, S. S. Kim, K. J. Yoo, H. Y. Park, Y. Jang, B. C. Chang, C. Y. Choi, K. C. Hwang and B. S. Kim, Ann. Surg., 2005, 241, 506–515 CrossRef.
- B. Tschoeke, T. C. Flanagan, S. Koch, M. S. Harwoko, T. Deichmann, V. Ella, J. S. Sachweh, M. Kellomaki, T. Gries, T. Schmitz-Rode and S. Jockenhoevel, Tissue Eng. A, 2009, 15, 1909–1918 Search PubMed.
- S. J. Lee, S. H. Oh, J. Liu, S. Soker, A. Atala and J. J. Yoo, Biomaterials, 2008, 29, 1422–1430 CrossRef CAS.
- Zhang, J. Y. Zhou, Q. P. Lu, Y. J. Wei and S. S. Hu, Biotechnol. Bioeng., 2008, 99, 1007–1015 CrossRef.
- M. Wilhelmi, S. Koenneker, M. Bonehie, M. Pflaum, S. Jockenhoevel and A. Haverich, Eur. J. Vasc. Endovasc. Surg., 2009 Search PubMed , submitted.
| This journal is © The Royal Society of Chemistry 2010 |