Synthesis and characterization of reactive PEO–PMCL polymersomes†
Matthew A.
Petersen
,
Ligeng
Yin
,
Efrosini
Kokkoli
and
Marc A.
Hillmyer
*
Department of Chemical Engineering and Materials Science and Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455-0431, USA. E-mail: kokkoli@umn.edu; hillmyer@umn.edu
First published on 22nd June 2010
Abstract
Targeted drug delivery offers the possibility to greatly improve treatment outcomes for diseases such as cancer by enhancing specificity, thus minimizing detrimental side effects from off target delivery. Polymer vesicles, or polymersomes, have been shown to be a promising delivery vehicle capable of encapsulating a variety of hydrophilic and hydrophobic drugs in a nanoscale, long-circulating carrier. We have synthesized and thoroughly characterized the end-functionalized and amphiphilic diblock polymer poly(ethylene oxide)-block-poly(γ-methyl-ε-caprolactone). This block copolymer self-assembles in water to form polymersomes containing the vinyl sulfone electrophile at the poly(ethylene oxide) corona terminus. Following polymersome self-assembly, this functional group underwent efficient, site-selective attachment to a thiol-containing targeting peptide by a conjugate addition reaction under mild conditions and did not react with targeting peptides lacking a thiol. The vesicle morphology was unaltered following peptide addition. This work develops a versatile platform for the targeted delivery of therapeutics.
Introduction
A central challenge in cancer therapy today is attaining specific, targeted delivery of chemotherapeutics to diseased cells in tumors. Though highly effective cancer-killing drugs are available, in many cases these agents are limited clinically by drawbacks including systemic toxicity, dose-limiting side effects, and an insufficient concentration of drug in the tumor itself for effective treatment.1,2 Delivery using self-assembled block polymers has been widely investigated as a potential route to minimizing nonspecific delivery. When dispersed in aqueous media amphiphilic block polymers minimize unfavorable solvent interactions with the hydrophobic block by forming supramolecular aggregates, the precise morphology of which can be understood as a balance between energy associated with core chain stretching, corona chain repulsion and interfacial tension between core blocks and solvent.3,4 Varying block lengths adjusts this balance and results in a variety of morphologies, the most common of which are spherical micelles, wormlike micelles, and vesicles. The last of these, termed polymersomes5 by analogy to small-molecule liposomes, are spheres from tens of nanometres to microns in diameter comprised of an aqueous core and a “bilayered” hydrophobic shell covered on either side by a dense corona of hydrophilic polymer.6Polymersomes offer a number of attractive features as drug delivery vehicles. The presence of both a hydrophilic core and a relatively thick hydrophobic shell (as compared to liposomes) enables simultaneous encapsulation of hydrophilic and hydrophobic payloads. Their sizes can be easily reduced following self-assembly by techniques such as sonication, freeze–thaw cycles and extrusion through appropriately sized membranes to the 100–200 nm scale.7 At this length scale the polymersomes can most effectively leverage the Enhanced Permeability and Retention (EPR) effect and leave the bloodstream at the site of tumors.8,9 Polymersomes also offer the advantages of prolonged circulation in vivo,10 stability against dissociation,11 and robust mechanical strength.12 In each of these respects polymersomes surpass the capabilities of currently available systems using sterically stabilized liposomes.6
A variety of amphiphilic polymers have been shown to form polymersomes, with early examples generally being non-degradable model systems5,13,14 and more recent efforts moving toward degradable or otherwise environmentally responsive systems.15 Among the most common of the latter is poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO–PCL).16–19 Hydrophilic, non-immunogenic PEO prolongs circulation times in the body due to its well-proven shielding capacity against protein opsonization.20 PCL is commonly used as a hydrophobic block due to its lack of toxicity21 and the fact that it undergoes hydrolysis at modest rates in physiological conditions yet degrades much more rapidly at the substantially lower pH encountered following endocytosis into cells.18 PCL, however, is a semicrystalline polymer, necessitating the use of either high temperatures or organic cosolvents to form polymersomes in water. This is a key drawback for drug delivery applications, as residual solvent may be toxic and high temperatures may be detrimental to fragile therapeutics. To address this concern, poly(γ-methyl-ε-caprolactone) (PMCL), a methyl-substituted and consequently amorphous form of PCL, has been investigated as an alternative hydrophobic block in PEO–PMCL diblock copolymers.22–24 Studies of PEO–PMCL have shown that these systems adopted morphologies akin to those in PEO–PCL at room temperature without cosolvent, forming a range of stable dispersions with morphologies ranging from spherical micelles to wormlike micelles to polymersomes depending on the weight fraction of the PMCL block.25
Actively targeted carriers have proven to be particularly promising for enhancing the specificity of drug delivery systems.26 By developing ligands specific to receptors overexpressed on the surface of diseased cells and tethering these ligands to the delivery vehicles it is possible to achieve specific binding between delivery vehicles and target cells.27 Biomimetic peptides are promising ligands for binding to target receptors with high specificity and affinity.28 Perhaps the most widely used targeting peptides in the literature are (arginine-glycine-aspartic acid) or RGD-containing sequences. The RGD sequence is derived from the 10th type III repeat of the extracellular membrane protein fibronectin and is a ligand for the integrin adhesion receptors.29,30 By more closely mimicking the structure of fibronectin's cell adhesion domain, RGD-based peptide ligands have been further developed to the point that they rival the binding of the native protein itself in terms of various cell adhesion phenomena.31,32
Past reports concerning vesicles formed from PEO–PMCL used diblock copolymers with no reactive chemical functionality at the ‘free’ methoxy terminus of the PEO block.25 As a result, attachment of an active targeting group such as an RGD-containing peptide to the exterior of a vesicle was not readily feasible. Replacing the unreactive methoxy end group with vinyl sulfone enables tethering of targeting groups between the polymersome surface (i.e., corona) and a free thiol such as that on the amino acid residue cysteine via conjugate addition under mild aqueous conditions.33–36 This strategy has recently been used to synthesize vinyl sulfone–PEO–poly(L-lactide) spherical micelles that were capable of reacting with thiols.37 Unlike carbodiimide-mediated condensation between a peptide amine and a carboxylic acid, this approach does not require harsh post-attachment deprotection to ensure attachment at the N-terminus of the peptide provided only a single thiol is present on the ligand.38 In contrast to ‘click’ reactions such as the copper-catalyzed Huisgen cycloaddition between azides and alkynes or the radical-initiated thiol–ene addition, a thiol–vinyl sulfone attachment strategy requires no toxic metal catalysts, no modification of the targeting ligand itself beyond inclusion of a cysteine residue, and no irradiation with ultraviolet radiation (for example in the thiol–ene click reaction) that may damage fragile therapeutics encapsulated in the carrier. Finally, unlike conjugate addition between thiols and maleimides, vinyl sulfone groups are less susceptible to addition of water across the double bond prior to conjugate addition,39 a key consideration given the intended application of the system. Incorporating vinyl sulfone functionality at the ‘free’ end of corona-forming PEO allows attachment of targeting molecules after self-assembly of vesicles, minimizing the potentially dramatic effects on carrier morphology of targeting moieties attached prior to self-assembly.40 In this way it is possible to use a variety of targeting ligands, peptide based or otherwise, without the need to verify the morphology of the carrier for each new case.
Herein we report the synthesis of a diblock copolymer capable of reacting with a targeting ligand under mild conditions via conjugate addition following vesicle formation. Following a related procedure by Bae et al. the polymer was synthesized by preferentially modifying one end of PEO diol with divinyl sulfone.37 After each synthetic step we carefully characterized all polymeric intermediates, allowing structures to be assigned for the desired singly modified PEO as well as side products. The remaining hydroxyl terminus was then used to initiate polymerization of MCL. Following isolation of the desired end-functional diblock copolymer (VS–PEO–PMCL), the polymer self-assembled in aqueous solution to form polymersomes analogous to those observed in unfunctionalized PEO–PMCL systems. These polymersomes could be modified with thiol-containing peptides by site-specific attachment to vinyl sulfone groups in the vesicle corona. Following peptide attachment the vesicle morphology was unchanged.
Experimental section
Materials
All chemicals were obtained from Sigma-Aldrich (St Louis, MO) and used as received unless otherwise noted. Water was obtained from a Milli-Q water system purified to a resistivity of 18.2 MΩ cm. Peptides were synthesized by the Oligonucleotide and Peptide Synthesis Facility at the University of Minnesota and their molar masses were confirmed by Matrix Assisted Laser Desorption Ionization Mass Spectrometry (MALDI-MS). 1H NMR spectra were acquired using a Varian VAC-300 spectrometer at room temperature. All polymer samples were dissolved in CDCl3 at approximately 1 w/v%. Chemical shifts were referenced to tetramethylsilane and integrations reported relative to that of 2.0 kg mol−1 PEO (180 methylene protons per chain). Size exclusion chromatography (SEC) was performed and polydispersity indices (PDIs) were determined using a Hewlett-Packard series 1100 liquid chromatography system equipped with a Hewlett-Packard 1047A RI detector and three PLgel 5 µm MIXED-C columns (polymer laboratories) with chloroform as the mobile phase (35 °C, 1 mL min−1) and calibrated using polystyrene standards (Polymer Laboratories). All phosphate buffered saline (PBS) was 100 mM, pH 7.5 with 5 mM ethylenediaminetetraacetic acid (EDTA). Dialysis bags were regenerated cellulose and purchased from Spectrum Labs. Fluorescence readings were acquired using a Varian Cary Eclipse fluorescence spectrophotometer.Mass spectrometry
Electrospray Ionization Mass Spectrometry (ESI-MS) samples were dissolved at approximately 1 w/v% in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 MeOH:H2O doped with approximately 0.1 w/v% KCl and measurements obtained using a Bruker Biotof II instrument in reflectron mode. MALDI-MS samples were dissolved at approximately 1 w/v% in THF containing 10 w/v% 2-(4-hydroxyphenylazo)-benzoic acid (HABA) and 1 w/v% NaCl as a cationizing agent followed by spotting of 0.7 µL on a MALDI target. Measurements were acquired using a Bruker Reflex III instrument in reflectron mode. Analysis and predictions for all MS data were performed using Bruker DataAnalysis software.
:50 MeOH:H2O doped with approximately 0.1 w/v% KCl and measurements obtained using a Bruker Biotof II instrument in reflectron mode. MALDI-MS samples were dissolved at approximately 1 w/v% in THF containing 10 w/v% 2-(4-hydroxyphenylazo)-benzoic acid (HABA) and 1 w/v% NaCl as a cationizing agent followed by spotting of 0.7 µL on a MALDI target. Measurements were acquired using a Bruker Reflex III instrument in reflectron mode. Analysis and predictions for all MS data were performed using Bruker DataAnalysis software.
Synthesis of non-reactive PEO–PMCL
MCL was prepared by Baeyer–Villiger oxidation of 4-methyl cyclohexanone using m-chloroperoxybenzoic acid (m-CPBA) as described elsewhere.24,25,41 PEO–PMCL was synthesized by ring opening polymerization of MCL from a PEO methyl ether (MePEO) macroinitiator with molecular weight 2 kg mol−1 catalyzed by SnOct2. In a sample reaction, MePEO (550 mg, 0.28 mmol) and a Teflon-coated stir bar were added to a previously flame-dried reaction vessel, heated to 110 °C under vacuum for 2 h, sealed and cooled to room temperature. In a dry box under inert atmosphere MCL (3.0 g, 23 mmol) and SnOct2 (12 µL, 37 µmol) were added. The reaction vessel was sealed, transferred to an oil bath and allowed to react at 110 °C for 1.5 h under mixing. The vessel, now containing a clear, viscous liquid, was then cooled to room temperature, dissolved in CH2Cl2, and precipitated twice into hexanes. 1H NMR (CDCl3): δ 4.10 (m, 142H, -C(O)-O-CH2-), 3.65 (s, 180H, -O-CH2-CH2-O-), 3.38 (s, 3H, CH3-O-), 2.30 (m, 142H, -CH2-C(O)-O-), 1.75–1.35 (m, 355H, -CH2-CH(CH3)-CH2-, -CH2-CH(CH3)-CH2-, and -CH2-CH(CH3)-CH2-), 0.92 (d, 213H, -CH2-CH(CH3)-CH3-). Mn 24.0 kg mol−1, PDI 1.19 (SEC).Synthesis of VS–PEO–PMCL
VS–PEO–PMCL synthesis was based on that of Bae.37 We have made significant modifications to enhance yields and provide precision characterization of the intermediates. The details are described below.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-SO2-), 6.46 (d, 0.85H, CH2CH-SO2-), 6.22 (d, 0.85H, CH2CH-SO2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 1.7H, -SO2-CH2-CH2-S-), 2.94 (m, 1.7H, -S-CH2-CH2-O-), 2.74 (t, 1.7H, -SO2-CH2-), 2.53 (bs, 1.3H, -OH); MALDI-MS: m/z unmodified PEO [M + Na+] calculated 2110.23, observed 2110.24; [M + K+] calculated 2126.21, observed 2126.22; VS–PEO [M + Na+] calculated 2112.14, observed 2112.18; [M + K+] calculated 2128.11, observed 2128.08; VS–PEO–VS [M + Na+] calculated 2114.05, observed 2114.04; [M + K+] calculated 2130.02, observed 2130.02. Mn 4.05 kg mol−1, PDI 1.13 (SEC).
CH-SO2-), 6.46 (d, 0.85H, CH2CH-SO2-), 6.22 (d, 0.85H, CH2CH-SO2-), 4.10 (m, 140H, -C(O)-O-CH2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 1.7H, -SO2-CH2-CH2-S-), 2.94 (m, 1.7H, -S-CH2-CH2-O-), 2.74 (t, 1.7H, -SO2-CH2-), 2.30 (m, 140H, -CH2-C(O)-O-), 1.75-1.35 (m, 350H, -CH2-CH(CH3)-CH2-, -CH2-CH(CH3)-CH2-, and -CH2-CH(CH3)-CH2-),.92 (d, 210H, -CH2-CH(CH3)-CH3-). Mn 29.3 kg mol−1, PDI 1.35 (SEC).
CH-SO2-), 6.46 (d, 0.8H, CH2CH-SO2-), 6.22 (d, 0.8H, CH2CH-SO2-), 4.10 (m, 140H, -C(O)-O-CH2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 2H, -SO2-CH2-CH2-S-), 2.94 (m, 2H, -S-CH2-CH2-O-), 2.74 (t, 2H, -SO2-CH2-), 2.30 (m, 140H, -CH2-C(O)-O-), 1.75-1.35 (m, 350H, -CH2-CH(CH3)-CH2-, -CH2-CH(CH3)-CH2-, and -CH2-CH(CH3)-CH2-), 0.92 (d, 210H, -CH2-CH(CH3)-CH3-, 210H). Mn 24.7 kg mol−1, PDI 1.11 (SEC).
CH-SO2-), 6.46 (d, 0.85H, CH2CH-SO2-), 6.22 (d, 0.85H, CH2CH-SO2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 1.7H, -SO2-CH2-CH2-S-), 2.94 (m, 1.7H, -S-CH2-CH2-O-), 2.74 (t, 1.7H, -SO2-CH2-), 2.53 (bs, 1.3H, -OH); MALDI-MS: m/z unmodified PEO [M + Na+] calculated 2110.23, observed 2110.24; [M + K+] calculated 2126.21, observed 2126.22; VS–PEO [M + Na+] calculated 2112.14, observed 2112.18; [M + K+] calculated 2128.11, observed 2128.08; VS–PEO–VS [M + Na+] calculated 2114.05, observed 2114.04; [M + K+] calculated 2130.02, observed 2130.02. Mn 4.05 kg mol−1, PDI 1.13 (SEC).
CH-SO2-), 6.46 (d, 0.85H, CH2CH-SO2-), 6.22 (d, 0.85H, CH2CH-SO2-), 4.10 (m, 140H, -C(O)-O-CH2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 1.7H, -SO2-CH2-CH2-S-), 2.94 (m, 1.7H, -S-CH2-CH2-O-), 2.74 (t, 1.7H, -SO2-CH2-), 2.30 (m, 140H, -CH2-C(O)-O-), 1.75-1.35 (m, 350H, -CH2-CH(CH3)-CH2-, -CH2-CH(CH3)-CH2-, and -CH2-CH(CH3)-CH2-),.92 (d, 210H, -CH2-CH(CH3)-CH3-). Mn 29.3 kg mol−1, PDI 1.35 (SEC).
CH-SO2-), 6.46 (d, 0.8H, CH2CH-SO2-), 6.22 (d, 0.8H, CH2CH-SO2-), 4.10 (m, 140H, -C(O)-O-CH2-), 3.65 (b, 180H, -O-CH2-CH2-O-), 3.31 (m, 2H, -SO2-CH2-CH2-S-), 2.94 (m, 2H, -S-CH2-CH2-O-), 2.74 (t, 2H, -SO2-CH2-), 2.30 (m, 140H, -CH2-C(O)-O-), 1.75-1.35 (m, 350H, -CH2-CH(CH3)-CH2-, -CH2-CH(CH3)-CH2-, and -CH2-CH(CH3)-CH2-), 0.92 (d, 210H, -CH2-CH(CH3)-CH3-, 210H). Mn 24.7 kg mol−1, PDI 1.11 (SEC).
Preparation of dispersions
All dispersions were prepared by thin film hydration. Polymer was dissolved in CH2Cl2 and added to a vial of known mass. Solvent was evaporated under an N2 stream to create a thin film then dried overnight at 45 °C under vacuum. PBS was then added to the sample to reach 1 w/v% polymer. A Teflon-coated stir bar was added, the vial sealed, and the solution stirred at room temperature for 1 week.Peptide attachment
Dispersions used for peptide attachment assays were prepared as described above and extruded through polycarbonate membranes of first 400 nm then 200 nm. Peptides were prepared as a stock solution in PBS. 1100 µL of dispersed polymer were transferred to a vial containing a Teflon-coated stir bar. The desired amount of peptide was added from a stock solution followed by buffer to reach 1300 µL total volume. Samples were allowed to react at room temperature for 18 h while stirring. Unincorporated peptide was then removed by dialysis against 3 × 3 litre volumes of PBS using dialysis membranes with a MWCO of 3.5 kDa over 72 h. The amount of attached peptide was determined by rapid injection and immediate, vigorous mixing of 60 µL of fluorescamine (10 mg mL−1 in acetone) into each purified sample, measuring fluorescence using λex = 390 nm and λem = 500 nm at a 90° angle relative to the excitation signal and comparing values to a calibration curve of known concentrations of peptide. Values measured for samples not reacted with peptide were used to determine background and subtracted from peptide-containing samples. All recorded fluorescence data are the average of ten fluorescence measurements (n = 10). Each experiment was performed in triplicate.Microscopy
Samples to be imaged by cryogenic transmission electron microscopy (cryo-TEM) were prepared using a home-built controlled environment vitrification system (CEVS) at room temperature.44 An aliquot of micelle solution (typically 8 µL) was loaded onto a lacey carbon support film, which was held by a pair of tweezers in the CEVS filled with saturated water vapor. Excess solution was blotted away using a piece of filter paper to form a thin film with thickness of approximately 100–300 nm spanning the holes of lacey carbon. About 15 seconds of waiting time was allowed to relax possible deformations formed during blotting. The grid was then quickly plunged into melting ethane (at ∼90 K) cooled by surrounding liquid nitrogen. The vitrified sample was then transferred onto Gatan 626 cryogenic sample holder, and examined in a JEOL 1210 TEM with accelerating voltage of 120 kV at about −177 °C. Images were collected using a Gatan 724 multiscan CCD and processed with Digital Micrograph version 3.3.1. Phase contrast was enhanced by imaging with 3–20 µm under focus.Results and discussion
The vinyl sulfone functional group has been employed to attach peptides to chain ends of non-degradable polymersome-forming systems.40 However, the somewhat harsh reaction conditions employed to install the vinyl sulfone moiety are not applicable to PMCL systems due to the susceptibility of this aliphatic polyester to degradation. It is therefore necessary to install the vinyl sulfone moiety prior to polymerization of MCL. The key step of such a synthesis is selective end-modification of a precursor PEO diol to allow installation of vinyl sulfone on one end of the molecule while maintaining the hydroxyl group necessary to initiate polymerization of MCL using ring-opening polymerization. The synthetic route to vinyl sulfone modified PEO is shown in Scheme 1.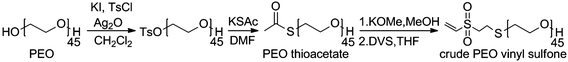 | ||
| Scheme 1 Idealized synthetic route to vinyl sulfone modified PEO (VS–PEO) from PEO diol. | ||
Bouzide and Sauvé developed an approach using silver(I) oxide to selectively monotosylate a variety of diols, among them oligo(ethylene glycols).45 The authors reported 85% monotosylation of hexaethylene glycol in the presence of a stoichiometric amount of tosyl chloride. This is a far more selective result than that obtained using a simple base-catalyzed reaction with a stoichiometric amount of tosyl chloride, which would be expected to yield a statistical 1:2:1 mixture of unmodified diol, the desired monotosylate and the bistosylate, respectively. Bouzide and Sauvé noted that as the length of polymer chain between end groups increased the selectivity of monotosylation decreased.45 This method of selective end-modification has since been applied to PEO with as many as 80 repeat units (3.5 kg mol−1),37,46 yet no quantification was performed with regard to selectivity in such large molecules. We address this issue below for PEO with molecular weight of 2 kg mol−1, qualitatively confirming the existence of all three forms of PEO by mass spectrometry and quantitatively estimating the relative fractions of each of the three products following polymerization of MCL.
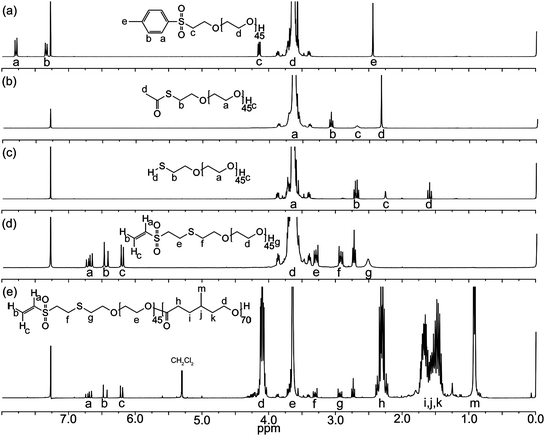
After silver(I) oxide-mediated tosylation of PEO, the resulting white solid was analyzed by 1H NMR spectroscopy, and we determined that 85% of PEO chains had a tosyl group installed, on average (Fig. 1a). ESI-MS measurements were consistent with the presence of the desired monotosylate (Ts–PEO). Overlapping peaks for unmodified PEO and the bistosylate (Ts–PEO–Ts) confirmed that at least one was present but made it impossible to confirm the presence of both of these polymers (Fig. S1, ESI†). The entire sample of tosylated polymer was reacted with an excess of potassium thioacetate in DMF to displace the tosyl groups with thioacetates. 1H NMR analysis confirmed that tosyl groups were displaced to form the thioacetate (Fig. 1b). The product was further characterized using MALDI-MS and compared to predictions for the monothioacetate as well as unmodified PEO and the AcS–PEO–AcS (Fig. 2). Molecular weights and isotopic abundances were consistent with theoretical predictions, confirming the coexistence of these PEO derivatives along with the desired heterotelechelic form of the polymer (AcS–PEO–OH).
 | ||
| Fig. 1 1H NMR spectra (CDCl3) of (a) PEO tosylate, (b) PEO thioacetate, (c) PEO thiolate, (d) PEO vinyl sulfonate and (e) VS–PEO–PMCL. | ||
 | ||
| Fig. 2 Predicted (a–c) and experimental (d) MALDI-MS data for a combination of unmodified PEO, AcS–PEO and AcS–PEO–AcS. The agreement between the predicted peaks and those observed in experimental measurements is consistent with a mixture of these three forms of PEO. | ||
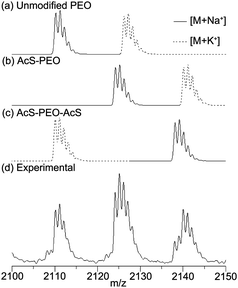
The mixture of thioacetates was deprotected to the corresponding thiols by dissolution in methanol containing potassium methoxide. The SEC trace of the products was multimodal (Fig. 3), due to the formation of disulfide bonds. Two cycles of reduction with aqueous sodium borohydride43 followed by extraction into an organic phase and precipitation into cold diethyl ether effectively reduced disulfide bonds. The SEC trace of this sample was nearly monomodal, consistent with reduction of disulfide links between chain ends. Following reduction, MALDI-MS data was consistent with the theoretical predictions for HS–PEO, PEO and the HS–PEO–SH, with distinct peaks apparent for each molecule (Fig. S2, ESI†) while 1H NMR spectroscopy confirmed complete conversion to the thiol (Fig. 1c). Reoxidation to the disulfide could be accomplished by bubbling air through the sample (Fig. 3).
 | ||
| Fig. 3 Thiol-modified PEO is prone to oxidation to form disulfide bonds both between ends and with thiol-based impurities. Cleavage of these disulfide bonds is essential both for purification and to ensure free thiols are available for reaction with divinyl sulfone. (a) Following cleavage of the thioacetate many PEO chains are linked via disulfide bonds, leading to a high PDI. (b) Following reduction only minimal low elution volume peak is visible and the polydispersity index is much lower, consistent with free chains. (c) Disulfide bonds can be reintroduced by bubbling air through the sample. | ||
Divinyl sulfone was added to thiol-modified end groups by conjugate addition between thiols and vinyl sulfone. To minimize disulfide bond formation all solvent was thoroughly degassed and reactions performed under nitrogen atmosphere. After dissolution of polymer, a small amount of dithiothreitol was added to reduce any fortuitously formed disulfide bonds. Divinyl sulfone was then added rapidly under vigorous stirring at large excess to minimize chain–chain coupling. Repeated precipitation into ice-cold diethyl ether yielded a white solid free of unattached divinyl sulfone with quantitative reaction of thiol groups as determined by 1H NMR spectroscopy (Fig. 1d). A small peak at low elution volume was detected by SEC and was unaffected by the addition of reductants, implying some divinyl sulfone mediated chain–chain coupling. Experimental MALDI-MS data were consistent with simulated predictions of VS–PEO, PEO, and VS–PEO–VS (Fig. 4). To verify that no reaction would occur between hydroxyl groups and vinyl sulfone, unmodified PEO was mixed with divinyl sulfone under identical conditions to those used to install vinyl sulfone on the thiol-terminated system. Following precipitation into cold diethyl ether no evidence of divinyl sulfone was observed in the 1H NMR spectrum, confirming minimal reactivity with hydroxyl groups under these conditions.
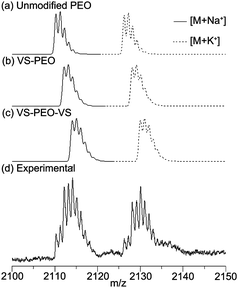 | ||
| Fig. 4 Predicted (a–c) and experimental (d) MALDI-MS data for a combination of unmodified PEO, VS–PEO and VS–PEO–VS. The agreement between the predicted peaks and those observed in experimental measurements is consistent with a mixture of these three forms of PEO. | ||
After installation of vinyl sulfone, hydroxyl end groups were used as initiation sites for polymerization of MCL. In past studies, PEO–PMCL has primarily been prepared in organic solvent using aluminium alkoxides and PEO monomethyl ether as the initiating system.23–25 Initial experiments using these conditions and vinyl sulfone modified PEO (VS–PEO) resulted in loss of vinyl sulfone groups. To address this concern, SnOct2 was employed as the catalyst and no solvent was used. No loss of vinyl sulfone was detectable by 1H NMR spectroscopy under these conditions. In all cases a diblock of approximately 11 kg mol−1 was targeted (i.e., PEO 2 kg mol−1, PMCL 9 kg mol−1), a molecular weight similar to that shown to form vesicles in studies of unreactive PEO–PMCL.25 When PEO methyl ether was used to initiate polymerization it was possible to obtain the unreactive diblock polymer PEO–PMCL while maintaining PDI values below 1.2 as long as the polymerizations were not allowed to proceed to full conversion. When vinyl sulfone modified PEO was used to initiate polymerization, polymerization proceeded from all available hydroxyl groups as shown in the SEC data presented in Fig. 5. Thus VS–PEO formed the desired vinyl sulfone-functionalized block polymer VS–PEO–PMCL. However, initiation also occurred at both ends of residual unmodified PEO to yield PMCL–PEO–PMCL (low elution volume). VS–PEO–VS remained as well (evident at high elution volumes). The agreement between predicted and experimental mass spectra for modified PEO above clearly suggested that some mixture of singly, doubly and unmodified PEO was present; however, quantification of the relative populations of each product by MALDI-MS is challenging47–49 and necessitated quantification by other means. By using 1H NMR data following polymerization of MCL we were able to determine that the mixture was composed of 10 mol% VS–PEO–VS (2270 g mol−1), 70 mol% VS–PEO–PMCL (11 kg mol−1) and 20 mol% PMCL–PEO–PMCL (ca. 20 kg mol−1) (ESI†).
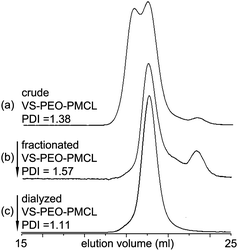 | ||
| Fig. 5 SEC elugrams showing the purification of VS–PEO–PMCL diblock. (a) Polymerization initiated from hydroxyl groups on vinyl sulfone modified PEO. Unmodified PEO formed the triblock PMCL–PEO–PMCL at low elution volumes, VS–PEO formed the diblock VS–PEO–PMCL and comprises the middle peak and VS–PEO–VS initiated no polymerization of MCL and was eluted last. (a) Following precipitation from CH2Cl2 to remove PMCL–PEO–PMCL the diblock VS–PEO–PMCL remained, as did VS–PEO–VS. (c) After dialysis only the desired VS–PEO–PMCL diblock remained. | ||
Following MCL polymerization fractional precipitation was used to isolate the target VS–PEO–PMCL diblock (Scheme 2), taking advantage of the relatively greater hydrophobicity of the triblock PMCL–PEO–PMCL. By slow addition of methanol to a dichloromethane solution of the crude mixture until the point of cloudiness and centrifuging the mixture it was possible to gradually adjust solvent quality to be incompatible with triblock polymer while leaving most of the diblock and nearly all of the VS–PEO–VS in solution (Fig. 5b). By formulating similar arguments to those used in to estimate the quantity of each polymer following MCL polymerization it was possible to estimate that 60 mol% of the remaining mixture of VS–PEO–PMCL and VS–PEO–VS after fractional precipitation was comprised of the VS–PEO–PMCL diblock. Dialysis against water was used to remove residual VS–PEO–VS. Following dialysis and lyophilization no degradation of the polymer chain was detectable by SEC (Fig. 5c) and only a small degree of vinyl sulfone loss was detectable by 1H NMR spectroscopy (∼10%, Fig. 1e), yielding the desired diblock as a viscous, transparent liquid.
 | ||
| Scheme 2 Polymerization of MCL from crude VS–PEO macroinitiator and purification used to isolate VS–PEO–PMCL diblock. | ||
To verify that aggregate morphology was unaffected by vinyl sulfone groups and attached peptide, cryo-TEM was used to compare the solution morphology of VS–PEO–PMCL without peptide (VS-0) and samples densely grafted with peptide (VS-100) to the control sample of unreactive PEO–PMCL. In the inert system (Fig. 6a) vesicles were the primary morphology observed. Some wormlike micelles were also present, consistent with coexisting morphologies observed in a number of polymer systems, including PEO–PMCL.25,50,51 When vinyl sulfone groups were present (sample VS-0, Fig. 6b) there was little discernible difference in morphology from that expected in the absence of a reactive group and only vesicles were observed.
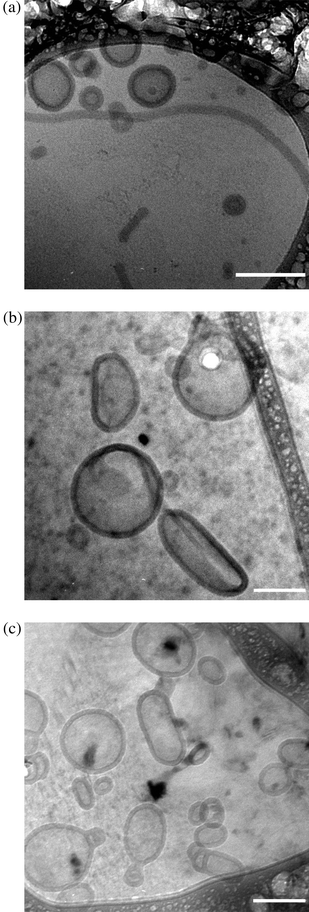 | ||
| Fig. 6 Cryo-TEM of aggregates formed by thin film hydration of the diblock copolymers: (a) Unreactive PEO–PMCL, (b) reactive VS–PEO–PMCL sans peptide (VS-0) and (c) reactive VS–PEO–PMCL with approximately 40 mol% of all vinyl sulfone groups (both exterior- and interior-facing) tethered to the thiol-bearing peptide CGRGDS (VS-100). In PEO–PMCL (a) vesicles are the predominant morphology with some coexisting wormlike micelles, consistent with prior studies of this system.25 In the VS–PEO–PMCL system prior to peptide attachment (b) vesicles are again the predominant morphology, with diameter consistent with extrusion through a 200 nm mesh. No visible change in morphology is observed upon tethering of a nearly 600 g mol−1 targeting moiety (CGRGDS) following self-assembly. Large black structures on the periphery of images are the lacey carbon used to suspend samples for vitrification. All scale bars 200 nm. | ||
To probe the capacity of this vinyl sulfone functional polymer to attach peptides, dispersions of either non-reactive PEO–PMCL (9100 g mol−1 PMCL, 18 wt% PEO, PDI 1.19) or reactive VS–PEO–PMCL (8900 g mol−1 PMCL, 18 wt% PEO, PDI 1.11) were formed at 1 w/v% in PBS (pH 7.5). Following extrusion to 200 nm, the desired amount of peptide (CGRGDS or GRGDS) was added to the polymersome solution and allowed to react for 18 hours then the solution was dialyzed against pure water. To determine the amount of peptide attached to polymersomes, the fluorescamine assay was used.52 A fluorescence-based assay was chosen due to the fact that the more common colorimetric assays rely on measures of absorbance to quantify peptide concentrations. In the case of polymersomes, solutions are not necessarily clear even prior to the addition of assay reagents; as a result, absorbance measurements are particularly susceptible to interference from the dispersion itself. The superior selectivity of the fluorescence-based assay (minimal fluorescence unless fluorescamine reacts with peptide amines) and 90° detection angle should decrease interference relative to absorbance-based assays even if the solution scatters light. In the present study, any interference from the solution would result in an underestimation of attached peptide, whereas an absorbance-based assay would overestimate this quantity.
Fluorescence values obtained for VS–PEO–PMCL dispersions without peptide were used as a measure of background (sample VS-0) and peptide amounts determined by comparison to fluorescence of calibration curves containing known amounts of peptide (Fig. S6, ESI†). The results of this assay are presented in Table 1. When peptides contained a thiol-bearing cysteine residue even low concentrations of peptide were highly reactive, and nearly all of the peptide coupled to vinyl sulfone groups on the surface of vesicles. As the amount of peptide was increased relative to the total vinyl sulfone groups present on both the exterior and interior interface the amount of coupled peptide gradually plateaued at slightly over 40% of all vinyl sulfone groups. This is consistent with only outwardly oriented vinyl sulfone groups being available for reaction with thiols, as approximately half of the polymer chains on a given aggregate are situated on the inner surface of the polymersome and thus inaccessible to peptides. As approximately 10% of vinyl sulfone groups were hydrolyzed during dialysis purification and the measured level of peptide attachment is consistent with saturation or near-saturation of remaining reactive sites. Any residual unreacted vinyl sulfone on the vesicle's exterior interface in high peptide-concentration samples (VS-100 and VS-500) are likely due to dense crowding following peptide attachment, resulting in steric restraints hindering the addition of further peptides.
To ensure proper function of targeting groups it is essential that the attachment of ligands not only proceeds to high reaction conversions but also be site-specific, as simply tethering a targeting group without regard for orientation will result in decreased ligand activity.53,54 Here presentation of the peptide with a specific orientation is facilitated by attachment solely due to reaction between the thiol on a cysteine residue at the N-terminus of the peptide and a vinyl sulfone group in the vesicle corona. Of particular concern is that amines can act as donors in conjugate additions. To ensure that peptide attachment was not due to the reaction of amines GRGDS peptide, which lacks a cysteine residue but contains a terminal amine, was reacted with VS–PEO–PMCL vesicles. Following dialysis only a small amount of peptide was detected (0.25 mol% ± 0.45%), suggesting that if peptide amines do react with vinyl sulfone groups, this reaction occurs much less readily than is the case for thiols, a finding consistent with prior reports about the relative reactivity of each functional group in conjugate addition.55 When cysteine-containing peptides were mixed with polymersomes lacking the vinyl sulfone groups (O-100) minimal peptide was detected (0.25 mol% ± 0.64%), implying that both a vinyl sulfone group and a thiol are required to be present for peptide attachment. These results bode well for preserving site selective attachment of targeting groups, as the vast majority of attachment will occur between a vinyl sulfone group in the polymersome corona and a peptide's N-terminal thiol. No change in morphology was observed following the near-saturation of the vesicle surface with peptide (sample VS-100, Fig. 6c).
Conclusions
To develop a degradable, targeted drug delivery system, we have synthesized, isolated and characterized the vesicle-forming block polymer VS–PEO–PMCL. This amphiphile self-assembles in water to form vesicles with reactive vinyl sulfone groups at the terminus of the corona-forming PEO block. These groups were capable of tethering high concentrations of a thiol-containing targeting peptide after self-assembly of vesicles in mild conditions without the need for any catalyst. Attachment of thiol-containing targeting peptides proceeded to high conversion while maintaining orientation of peptide (CGRGDS) while peptides lacking a thiol group (GRGDS) underwent minimal attachment. Following targeting peptide attachment no change was observed in aggregate morphology. Previous in vitro studies have shown that even low concentrations of peptides can dramatically enhance binding and endocytosis of functionalized vesicles (polymersomes or stealth liposomes) by cells overexpressing the targeted receptors.38,56,57 We anticipate that the similarity between PEO–PMCL and VS–PEO–PMCL will allow these polymers to be homogenously blended. By including VS–PEO–PMCL as the minority component prior to self-assembly it should be possible to simultaneously take advantage of both the shielding capacity of the PEO corona and the enhanced targeting due to ligand bound to vinyl sulfone groups on the exterior of the vesicles. This system should serve as a versatile platform for specific delivery of a variety of therapeutic payloads in a carrier capable of utilizing a wide range of targeting moieties.Acknowledgements
We would like to thank Todd O. Pangburn for suggesting the use of the flourescamine assay for peptide quantification, Brett Waybrant for help in mass spectrometry, and Alex Madsen for assistance in synthesis. This work was supported primarily by the MRSEC Program of the National Science Foundation under award number DMR-0212302 and DMR-0819885. Part of this work was carried out in the Institute of Technology Characterization Facility, University of Minnesota, which has received capital equipment funding from the NSF through the MRSEC, ERC and MRI programs, and the Department of Chemistry Molecular Characterization Facilities, University of Minnesota, which have received capital equipment funding from the NSF through the MRSEC.References
- M. Majer, W. Akerley and S. K. Kuwada, Oncologists' current opinion on the treatment of colon carcinoma, Anti-Cancer Agents Med. Chem., 2007, 7, 492–503 CAS.
- A. H. Partridge and E. P. Winer, Long-term complications of adjuvant chemotherapy for early stage breast cancer, Breast Dis., 2004, 21, 55–64 Search PubMed.
- A. Halperin, M. Tirrell and T. Lodge, Tethered Chains in Polymer Microstructures, in Macromolecules: Synthesis, Order and Advanced Properties, 1992 ed. A. Abe, H. Benoit, H.-J. Cantow, P. Corradini, K. Dušek, S. Edwards, H. Fujita, G. Glöckner, H. Höcker, H.-H. Hörhold, H.-H. Kausch, J. P. Kennedy, J. L. Koenig, A. Ledwith, L. Monnerie, S. Okamura, C. G. Overberger, H. Ringsdorf, T. Saegusa, J. C. Salamone, J. L. Schrag and G. Wegner, Springer-Verlag, pp. 31–71 Search PubMed.
- A. E. Lifeng Zhang, Formation of crew-cut aggregates of various morphologies from amphiphilic block copolymers in solution, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Polymersomes: tough vesicles made from diblock copolymers, Science, 1999, 284, 1143–1146 CrossRef.
- D. E. Discheri and F. Ahmed, Polymersomes, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS.
- J. C.-M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y.-Y. Won, F. S. Bates and D. E. Discher, Preparation, stability, and in vitro performance of vesicles made with diblock copolymers, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef CAS.
- M. Gaumet, A. Vargas, R. Gurny and F. Delie, Nanoparticles for drug delivery: the need for precision in reporting particle size parameters, Eur. J. Pharm. Biopharm., 2008, 69, 1–9 CrossRef CAS.
- S. Unezaki, K. Maruyama, J.-I. Hosoda, I. Nagae, Y. Koyanagi, M. Nakata, O. Ishida, M. Iwatsuru and S. Tsuchiya, Direct measurement of the extravasation of polyethyleneglycol-coated liposomes into solid tumor tissue by in vivo fluorescence microscopy, Int. J. Pharm., 1996, 144, 11–17 CrossRef CAS.
- P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, Polymer vesicles in vivo: correlations with PEG molecular weight, J. Controlled Release, 2003, 90, 323–334 CrossRef.
- Y. Y. Won, H. T. Davis and F. S. Bates, Molecular exchange in PEO–PB micelles in water, Macromolecules, 2003, 36, 953–955 CrossRef CAS.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Molecular weight dependence of polymersome membrane structure, elasticity, and stability, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- K. Yu, L. Zhang and A. Eisenberg, Novel morphologies of “crew-cut” aggregates of amphiphilic diblock copolymers in dilute solution, Langmuir, 1996, 12, 5980–5984 CrossRef CAS.
- L. Zhang and A. Eisenberg, Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers, Science, 1995, 268, 1728–1731 CrossRef CAS.
- F. Meng, Z. Zhong and J. Feijen, Stimuli-responsive polymersomes for programmed drug delivery, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Shrinkage of a rapidly growing tumor by drug-loaded polymersomes: pH-triggered release through copolymer degradation, Mol. Pharmaceutics, 2006, 3, 340–350 CrossRef CAS.
- Y. Geng and D. E. Discher, Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles, J. Am. Chem. Soc., 2005, 127, 12780–12781 CrossRef CAS.
- F. Ahmed and D. E. Discher, Self-porating polymersomes of PEG–PLA and PEG–PCL: hydrolysis-triggered controlled release vesicles, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- P. P. Ghoroghchian, G. Li, D. H. Levine, K. P. Davis, F. S. Bates, D. A. Hammer and M. J. Therien, Bioresorbable vesicles formed through spontaneous self-assembly of amphiphilic poly(ethylene oxide)-block-polycaprolactone, Macromolecules, 2006, 39, 1673–1675 CrossRef CAS.
- L. Van Vlerken, T. Vyas and M. Amiji, Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS.
- M. S. Taylor, A. U. Daniels, K. P. Andriano and J. Heller, Six bioabsorbable polymers: in vitro acute toxicity of accumulated degradation products, J. Appl. Biomater., 1994, 5, 151–157 Search PubMed.
- P. Vangeyte, B. Leyh, M. Heinrich, J. Grandjean, C. Bourgaux and R. Jerome, Self-assembly of poly(ethylene oxide)-b-poly(ε-caprolactone) copolymers in aqueous solution, Langmuir, 2004, 20, 8442–8451 CrossRef CAS.
- P. Vangeyte, S. Gautier and R. Jerome, About the methods of preparation of poly(ethylene oxide)-b-poly(ε-caprolactone) nanoparticles in water: analysis by dynamic light scattering, Colloids Surf., A, 2004, 242, 203–211 CrossRef CAS.
- P. Vangeyte and R. Jérôme, Amphiphilic block copolymers of high-molecular-weight poly(ethylene oxide) and either ε-caprolactone or γ-methyl-ε-caprolactone: synthesis and characterization, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1132–1142 CrossRef CAS.
- J. A. Zupancich, F. S. Bates and M. A. Hillmyer, Aqueous dispersions of poly(ethylene oxide)-b-poly(γ-methyl-ε-caprolactone) block copolymers, Macromolecules, 2006, 39, 4286–4288 CrossRef.
- T. O. Pangburn, M. A. Petersen, B. Waybrant, M. M. Adil and E. Kokkoli, Peptide- and aptamer-functionalized nanovectors for targeted delivery of therapeutics, J. Biomech. Eng., 2009, 131, 074005 CrossRef.
- T. Minko, S. S. Dharap, R. I. Pakunlu and Y. Wang, Molecular targeting of drug delivery systems to cancer, Curr. Drug Targets, 2004, 5, 389–406 Search PubMed.
- E. Kokkoli, A. Mardilovich, A. Wedekind, E. L. Rexeisen, A. Garg and J. A. Craig, Self-assembly and applications of biomimetic and bioactive peptide-amphiphiles, Soft Matter, 2006, 2, 1015–1024 RSC.
- M. D. Pierschbacher and E. Ruoslahti, Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule, Nature, 1984, 309, 30–33 CrossRef CAS.
- E. Ruoslahti and M. Pierschbacher, New perspectives in cell adhesion: RGD and integrins, Science, 1987, 238, 491–497 CrossRef CAS.
- J. A. Craig, E. L. Rexeisen, A. Mardilovich, K. Shroff and E. Kokkoli, Effect of linker and spacer on the design of a fibronectin-mimetic peptide evaluated via cell studies and AFM adhesion forces, Langmuir, 2008, 24, 10282–10292 CrossRef CAS.
- A. Mardilovich, J. A. Craig, M. Q. Mccammon, A. Garg and E. Kokkoli, Design of a novel fibronectin-mimetic peptide-amphiphile for functionalized biomaterials, Langmuir, 2006, 22, 3259–3264 CrossRef CAS.
- S. C. Rizzi and J. A. Hubbell, Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrixes. Part I: development and physicochemical characteristics, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS.
- M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Systematic modulation of Michael-type reactivity of thiols through the use of charged amino acids, Bioconjugate Chem., 2001, 12, 1051–1056 CrossRef CAS.
- A. B. Pratt, F. E. Weber, H. G. Schmoekel, R. Müller and J. A. Hubbell, Synthetic extracellular matrices for in situ tissue engineering, Biotechnol. Bioeng., 2004, 86, 27–36 CrossRef CAS.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Cell-responsive synthetic hydrogels, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- J. W. Bae, E. Lee, K. M. Park and K. D. Park, Vinyl sulfone-terminated PEG–PLLA diblock copolymer for thiol-reactive polymeric micelle, Macromolecules, 2009, 42, 3437–3442 CrossRef CAS.
- D. Demirgöz, T. O. Pangburn, K. P. Davis, S. Lee, F. S. Bates and E. Kokkoli, PR_b-targeted delivery of tumor necrosis factor-α by polymersomes for the treatment of prostate cancer, Soft Matter, 2009, 5, 2011–2019 RSC.
- M. Morpurgo, F. M. Veronese, D. Kachensky and J. M. Harris, Preparation and characterization of poly(ethylene glycol) vinyl sulfone, Bioconjugate Chem., 1996, 7, 363–368 CrossRef CAS.
- J. A. Zupancich, F. S. Bates and M. A. Hillmyer, Synthesis and self-assembly of RGD-functionalized PEO–PB amphiphiles, Biomacromolecules, 2009, 10, 1554–1563 CrossRef CAS.
- M. Trollsas, M. A. Kelly, H. Claesson, R. Siemens and J. L. Hedrick, Highly branched block copolymers: design, synthesis, and morphology, Macromolecules, 1999, 32, 4917–4924 CrossRef.
- D. E. Janssen and C. V. Wilson, Org. Synth., 1963, Coll. Vol. IV, 547.
- W. D. Brown, Reduction of protein disulfide bonds by sodium borohydride, Biochim. Biophys. Acta, 1960, 44, 365–367 CAS.
- J. R. Bellare, H. T. Davis, L. E. Scriven and Y. Talmon, Controlled environment vitrification system: an improved sample preparation technique, J. Electron Microsc. Tech., 1988, 10, 87–111 Search PubMed.
- A. Bouzide and G. Sauvé, Silver(I) oxide mediated highly selective monotosylation of symmetrical diols. Application to the synthesis of polysubstituted cyclic ethers, Org. Lett., 2002, 4, 2329–2332 CrossRef CAS.
- D. Shenoy, W. Fu, J. Li, C. Crasto, G. Jones, C. Dimarzio, S. Sridhar and M. Amiji, Surface functionalization of gold nanoparticles using hetero-bifunctional poly(ethylene glycol) spacer for intracellular tracking and delivery, Int. J. Nanomed., 2006, 1, 51–57 CrossRef CAS.
- S. J. Wetzel, C. M. Guttman and J. E. Girard, The influence of matrix and laser energy on the molecular mass distribution of synthetic polymers obtained by MALDI-TOF-MS, Int. J. Mass Spectrom., 2004, 238, 215–225 Search PubMed.
- G. Montaudo, F. Samperi and M. S. Montaudo, Characterization of synthetic polymers by MALDI-MS, Prog. Polym. Sci., 2006, 31, 277–357 CrossRef CAS.
- I. Schnöll-Bitai, T. Hrebicek and A. Rizzi, Towards a quantitative interpretation of polymer distributions from MALDI-TOF spectra, Macromol. Chem. Phys., 2007, 208, 485–495 CrossRef.
- S. Jain and F. S. Bates, Consequences of nonergodicity in aqueous binary PEO–PB micellar dispersions, Macromolecules, 2004, 37, 1511–1523 CrossRef CAS.
- S. Jain and F. S. Bates, On the origins of morphological complexity in block copolymer surfactants, Science, 2003, 300, 460–464 CrossRef CAS.
- S. Udenfriend, S. Stein, P. Böhlen, W. Dairman, W. Leimgruber and M. Weigele, Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range, Science, 1972, 178, 871–872 CAS.
- Y. Dori, H. Bianco-Peled, S. K. Satija, G. B. Fields, J. B. Mccarthy and M. Tirrell, Ligand accessibility as means to control cell response to bioactive bilayer membranes, J. Biomed. Mater. Res., 2000, 50, 75–81 CrossRef CAS.
- A. Mardilovich and E. Kokkoli, Biomimetic peptide-amphiphiles for functional biomaterials: the role of GRGDSP and PHSRN, Biomacromolecules, 2004, 5, 950–957 CrossRef CAS.
- M. Friedman, J. F. Cavins and J. S. Wall, Relative nucleophilic reactivities of amino groups and mercaptide ions in addition reactions with α, β-unsaturated compounds, J. Am. Chem. Soc., 1965, 87, 3672–3682 CrossRef CAS.
- D. Demirgöz, A. Garg and E. Kokkoli, PR_b-targeted PEGylated liposomes for prostate cancer therapy, Langmuir, 2008, 24, 13518–13524 CrossRef CAS.
- A. Garg, A. W. Tisdale, E. Haidari and E. Kokkoli, Targeting colon cancer cells using PEGylated liposomes modified with a fibronectin-mimetic peptide, Int. J. Pharm., 2009, 366, 201–210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MS, SEC and fluorescence calibration curve data for synthesis of VS–PEO–PMCL, PEO–PMCL and quantification of peptide attachment. See DOI: 10.1039/c0py00143k |
| This journal is © The Royal Society of Chemistry 2010 |