Novel aromatic polyamides with main chain and pendant 1,2,4-triazole moieties and their application to the extraction/elimination of mercury cations from aqueous media
Ana
Gómez-Valdemoro
,
Noelia
San-José
,
Félix Clemente
García
,
José Luis
De La Peña
,
Felipe
Serna
and
José Miguel
García
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Plaza de Misael Bañuelos s/n, 09001, Burgos, Spain. E-mail: jmiguel@ubu.es
First published on 15th June 2010
Abstract
This work describes five novel aromatic polyamides and copolyamides containing the triazole heterocycle attached by an amine linkage to the main and lateral chains. The polyamides were found to be amorphous and soluble in polar aprotic solvents. They demonstrated a film-forming capability, exhibited good thermal resistance and had high thermal transition temperatures (up to 375 °C). The triazole and amine groups imparted hydrophilicity to the polymers, giving rise to materials that improve their mechanical properties upon water uptake, and were efficient chelating/host units for heavy metal cations. These properties facilitated the preparation of solid polymer phases to be used for the extraction and elimination of environmentally deleterious cations from water environments. In regards to cation extraction from aqueous media with polyamide solid phases, the correct polymer structure designs led to the extraction of high percentages of HgII (neat 100%).
Introduction
The outstanding thermal and mechanical resistance of wholly aromatic polyamides (aramids) classifies them into the category of high-performance organic materials. Thus, the commercial aromatic polyamides, poly(p-phenylene terphthalamide), poly(m-phenylene isophthalamide), and copoly(p-phenylene/3,4′-oxydiphenylene terephthalamide), are useful polymers applied in advanced technologies, such as flame-resistant and high tensile strength coatings and fibres to be used as fabrics and fillers in the aerospace and armaments industry, in asbestos substitutes, as electrical insulation, in bullet-proof body armour, as protective clothing, industrial filters, and sports fabrics. Moreover, they are increasingly being used as advantageous replacements for metals or ceramics in currently produced goods, or even as new materials in novel technological applications.1–3Their properties arise from their aromatic structure and amide linkages, which result in stiff, rod-like macromolecular chains that interact with each other via strong and highly directional hydrogen bonds. These bonds create effective crystalline microdomains, resulting in high-level intermolecular packing and cohesive energy. As a result, they exhibit extremely high transition temperatures that lie above their decomposition temperatures, are sparingly soluble in common organic solvents and, accordingly, can only be transformed upon solution.
Research efforts are therefore underway to take advantage of these properties, enhance their processability and solubility, and incorporate new chemical functionalities in the polyamide backbone or in the lateral structure, so that their applicability is expanded and remains on the forefront of scientific research.1 Among others, the heterocycles are interesting chemical structures to modify the properties of aramids and other thermally stable polymers because they usually impart excellent thermal stability with improved solubility. As a consequence, research on aromatic polyamides with heterocycles in the main chain (oxadiazole,4 pyridine,5 diquinoline,6 phthalazinone,7 triazine,8 imide,9 benzofuran,10etc.) and in the pendant structure (carbazol,11 imidazole,12 benzoxazole,13 pyridine,14etc.) is widespread in the scientific literature.
Furthermore, the heterocycle properties and characteristics are expected to be retained by the polymers containing them. In this regard, the triazole derivatives are well-known binding sites for metals, especially for HgII, and applications of these kind of molecules for the removal of mercury cations from aqueous media, as well as sensing and recognition of mercury ions and the preparation of fluorescent on-off switchable chemosensors, have been described.15–17 Thus, the inclusion of the triazole heterocycle into macromolecular chains may permit the preparation of polymers with good mechanical, thermal and chemical properties that can be transformed into materials capable of interacting selectively with cations, specifically mercury ions. These polyamides could then be useful in the development of sensing materials, permselective membranes and solid phases to extract environmentally polluting cations, among other applications.
We have been working recently on the development of new aromatic and aliphatic-aromatic polyamides with a la carte properties, such as enhanced solubility, superior thermal and mechanical properties, high water uptake, recognition and sensing capabilities, and environmentally harmful cation extraction capabilities.18,19 The introduction of some kind of specific chemical functionality to the main or lateral chain can broaden the scope of the technological applications of the aromatic polyamides, giving rise to polymers with speciality properties, such as optical activity, electrochromicity and luminescence, special materials for membranes, and environmental or supramolecular-related technologies and applications.1,2
Continuing with our line of research, we describe in this work the synthesis and characterisation of five novel aromatic polyamides and copolyamides with 1,2,4-triazole moieties in the main and lateral structures. The polymers have been prepared by the polymerisation of two kinds of new monomers: aromatic diacids (A-A monomers), and an amino acid (A-B monomer). The properties of the polymers, in terms of thermal and mechanical behaviour, water uptake and solubility, have been analysed and a comparison with commercial aromatic polyamides is presented.
Moreover, the behaviour of the pendant or main chain triazole heterocycle as a heavy-metal chelating group has given rise to the development of polyamide solid phases with affinity toward heavy-metal cations, especially HgII, in aqueous environments. As a result, we have studied the ability of these materials to eliminate environmentally toxic heavy metal cations as their nitrate salts from aqueous media. The choice of the polyamide backbone for extraction experiments comes from the outstanding thermal and mechanical stability of this type of polymers, their hydrophilicity (necessary to extract analytes from water media), as well as their poor solubility in water and in common organic solvents, which makes them especially useful as solid materials in molecular recognition because they can be regenerated and reused by a simple filtration and washing procedure.1,19h–k
Experimental
All materials and solvents were commercially available and were used as received, unless otherwise indicated. N-Methyl-2-pyrrolidone (NMP, Aldrich) was vacuum-distilled twice over phosphorous pentoxide, and then stored over 4 Å molecular sieves. Lithium chloride (Aldrich) was dried at 400 °C for 12 h prior to use. Triphenylphosphite (TPP, Aldrich) was vacuum distilled twice over calcium hydride, and then stored over 4 Å molecular sieves. Pyridine (Merck) was dried at reflux temperature over sodium hydroxide for 24 h, and then distilled and stored over 4 Å molecular sieves. m-Phenylenediamine (MPD) and p-phenylenediamine (PPD) were commercially available (Aldrich) and were purified by double vacuum sublimation.Intermediates and monomers
The two aromatic diacids were synthesised following the reaction conditions described in Scheme 1.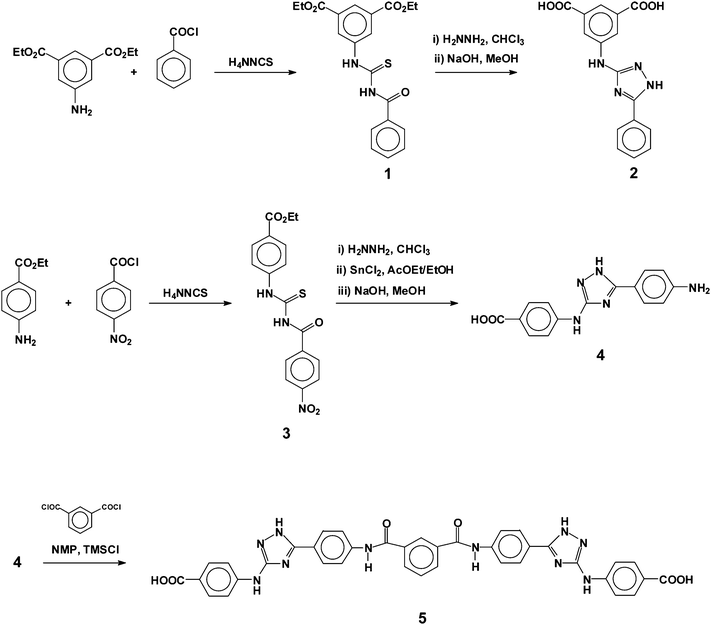 | ||
| Scheme 1 Monomer synthesis and labels. | ||
Yield: 90%. M.p.: 148 °C. 1H-NMR, δH (400 MHz, DMSO-d6, Me4Si): 12.71 (1H, s, NH); 11.76 (1H, s, NH); 8.52 (2H, s, Ph); 8.34 (1H, s, Ph); 8.03 (2H, d, J 7.2, Ph); 7.72–7.68 (1H, m, Ph); 7.59–7.56 (2H, m, Ph); 4.39 (4H, q, J 7.2, CH3CH2); 1,38 (6H, t, J 7.2, CH3CH2). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 180.85, 169.05, 165.41, 140.09, 133.22, 133.02, 131.73, 130.29, 129.75, 129.45, 127.83, 62.44, 15.11. EI-LRMS m/z: 400 (M+, 14), 237 (24), 192 (16), 105 (100), 77 (66). FTIR [wavenumbers (cm−1)]: νN–H: 3397; νC–H(s, as): 2985, 2902; νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 1722, 1670; δN–H: 1573; νAr CC: 1529; νCS: 1309; νC–O: 1240.
O: 1722, 1670; δN–H: 1573; νAr CC: 1529; νCS: 1309; νC–O: 1240.
Yield: 72%. M.p.: 228 °C. 1H-NMR, δH (400 MHz, DMSO-d6, Me4Si): 13.99 (1H, s, NH); 9.89 (1H, s, NH); 8.59 (2H, s, Ph); 8.04–8.00 (3H, m, Ph); 7.60–7.53 (3H, m, Ph); 4.40 (4H, J 7.2, CH3CH2); 1.39 (6H, t, J 7.2, CH3CH2). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 166.210, 161.10, 153.39, 143.81, 131.84, 131.05, 129.97, 128.07, 126.69, 121.18, 120.85, 61.96, 15.10. EI-LRMS m/z: 380 (M+, 100), 335 (14), 104 (45), 77 (16). FTIR [wavenumbers (cm−1)]: νN–H: 3341; νC–H(s, as): 2980, 2904; νCO:1722; δN–H:1612; νAr CC:1590; νCN:1549; νC–N:1468; νC–O: 1236.
In a 1 L Erlenmeyer flask fitted with a condenser and magnetic stirring, 10 mmol of 5-(5-phenyl-4H-1,2,4-triazol-3-ylamino)isophthalate was dissolved in 300 mL of methanol. About 75 mL of 20% aqueous sodium hydroxide was added and the mixture was then refluxed for 3 h, after which some methanol was removed under reduced pressure. 10% aqueous HCl solution was added drop wise to the mixture to adjust to pH 3 to precipitate 5-(5-phenyl-4H-1,2,4-triazol-3-ylamino)isophthalic acid (2). The product was filtered off, washed thoroughly with distilled water and MeOH, and dried in a vacuum oven overnight at 120 °C.
Yield: 92%. M.p.: 338 °C (d). 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 13.43 (3H, br s, NH, COOH); 9.87 (1H, NH); 8.54 (2H, s, Ph); 8.00–8.03 (3H, m, Ph); 7.59–7.50 (3H, m, Ph). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 168.01, 160.32, 154.34, 143.57, 132.74, 130.98, 130.07, 128.83, 126.81, 121.87, 121.41. EI-LRMS m/z: 324 (M+, 100), 181 (9), 119 (14), 104 (20), 77 (9). FTIR [Wavenumbers (cm−1)]: νN–H: 3328; νacid O–H: broadband (3204, 2812); νCO:1712; δN–H:1608; νAr CC:1590; νCN:1549; νC–N:1484.
Yield: 95%. M.p.: 179 °C. 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 12.61 (1H, s, NH); 9.25 (1H, s, NH); 8.38 (2H, d, J 8.8, Ph); 8.10–8.07 (4H, m, Ph); 7.86–7.84 (2H, J 8.8, Ph); 4.57 (2H, q, J 7.2, CH2CH3); 1.39 (3H, t, J 7.2, CH2CH3). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 177.63, 166.00, 165.38, 151.05, 141.43, 137.20, 130.74, 129.19, 128.97, 124.62, 123.28, 61.45, 14.60. EI-LRMS m/z: 373 (M+, 59), 179 (19), 165 (20), 150 (100), 120 (51), 104 (48). FTIR [Wavenumbers (cm−1)]: νN–H: 3338; νC–H(s, as): 2985, 2896; νCO: 1723, 1693; δN–H: 1604; νAr CC: 1597; νCS: 1309; νNO2 (s, as): 1534, 1347; νC–O: 1242.
Yield: 30%. M.p.: 310 °C. 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 14.00 (1H, s, NH); 10.05 (1H, s, NH); 8.38 (2H, d, J 8.4, Ph); 8.25 (2H, d, J 8.4, Ph); 7.92 (2H, d, J 8.4, Ph); 7.73 (2H, d, J 8.4, Ph); 4.29 (2H, q, J 7.2, CH2CH3), 1.33 (3H, t, J 7.2, CH2CH3). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 166.58, 157.11, 155.37, 148.45, 146.53, 136.75, 131.51, 127.65, 125.19, 121.78, 116.41, 61.08, 15.28. EI-LRMS m/z: 353 (M+, 100), 308 (49), 279 (4), 262 (15), 234 (5). FTIR [Wavenumbers (cm−1)]: νN–H: 3341; νC–H(s, as): 2985, 2903; νCO:1672; δN–H:1610; νAr CC:1591; νCN:1549; νC–N:1484; νNO2 (s, as): 1531, 1338; νC–O: 1283.
In a 250 mL Erlenmeyer flask fitted with magnetic stirring and a condenser, 10 mmol of ethyl 4-(5-(4-nitrophenyl)-4H-1,2,4-triazol-3-ylamino)benzoate, 50 mmol of SnCl2, 70 mL of ethyl acetate and 50 mL of ethanol were stirred together and heated under reflux. When the reaction mixture was completely dissolved, the reaction was kept at reflux for 2 h. Afterwards, solvents were removed under reduced pressure and the remaining solid was stirred with 150 mL of water. The pH was then made slightly basic (pH 7–8) by addition of 5% aqueous sodium bicarbonate. The resulting solid was filtered off and washed with water. The product, ethyl 4-(5-(4-aminophenyl)-4H-1,2,4-triazol-3-ylamino)benzoate, was extracted with methanol for 24 h in a Soxhlet.
Yield: 83%. M.p.: 241 °C. 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 13.40 (1H, br s, NH); 9.78 (1H, s, NH); 7.0 (2H, d, J 8.8, Ph); 7.73–7.70 (4H, m, Ph); 6.70 (2H, d, J 8.4, Ph); 5.63 (2H, s, NH); 4.29 (2H, q, J 7.2, CH2CH3); 1.33 (3H, t, J 7.2, CH2CH3). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 170.69, 164.45, 15.67, 155.60, 151.61, 135.44, 132.18, 124.61, 119.83, 119.49, 118.51, 64.90, 19.28. EI-LRMS m/z: 323 (M+, 100), 295 (13), 278 (35), 119 (33), 118 (24), 92 (8). FTIR [Wavenumbers (cm−1)]: νN–H: 3367; νC–H(s, as): 2978, 2903; νCO:1681; δN–H:1607; νAr CC:1587; νCN:1546; νC–O: 1279.
4-(5-(4-Aminophenyl)-4H-1,2,4-triazol-3-ylamino)benzoic acid (4) from 4-(5-(4-aminophenyl)-4H-1,2,4-triazol-3-ylamino)benzoate was prepared and purified in a similar manner to that of 5-(5-phenyl-4H-1,2,4-triazol-3-ylamino)isophthalic acid (2).
Yield: 89%. M.p.: 345 °C (d). 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 12.83 (1H, br s, COOH); 9.75 (1H, s, NH); 7.91 (2H, d, J 8.0, Ph); 7.75 − 7.71 (4H, m, Ph); 6.75 (2H, d, J 8.4, Ph), 5.96 (2H, br s, NH). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 168.42, 167.24, 160.16, 15.87, 150.98, 147.67, 147.37, 131.75, 131.59, 128.27, 121.59, 120.45, 116.20, 115.94, 115.85, 114.98. EI-LRMS m/z: 295 (M+, 100), 278 (2), 250 (4), 133 (8), 119 (38), 118 (27), 104 (5), 92 (7), 77 (6). FTIR [Wavenumbers (cm−1)]: νN–H: 3373; νacid O–H: broadband (3070, 2781); νCO:1679; δN–H:1613; νAr CC: 1586; νCN: 1554.
Yield: 98%. 1H-NMR δH (400 MHz, DMSO-d6, Me4Si): 13.85 (2H, br s, NH); 10.73 (2H, br s, NH); 9.88 (2H, br s, NH); 8.62 (1H, s, Ph); 8.23 (2H, d, J 7.6, Ph); 8.03 (8H, m, Ph); 7.89 (4H, d, J 9.2, Ph); 7.78 (1H, t, J 7.6, Ph); 7.70 (4H, d, J 8.8, Ph). 13C-NMR, δC (100.6 MHz, DMSO-d6, Me4Si): 171.09, 169.18, 167.15, 166.24, 166.11, 159.92, 154.35, 147.37, 146.56, 144.43, 135.99, 135.86, 131.96, 131.66, 129.76, 128.14, 127.42, 124.43, 123.17 121.38, 116.03, 115.84. FTIR [Wavenumbers (cm−1)]: νN–H: 3301; νacid O–H: broadband (3045, 2844); νCO:1681, 1650; δN–H:1607; νAr CC:1587; νCN:1537.
Polymer synthesis
The polymer synthesis and acronyms are depicted in Scheme 2.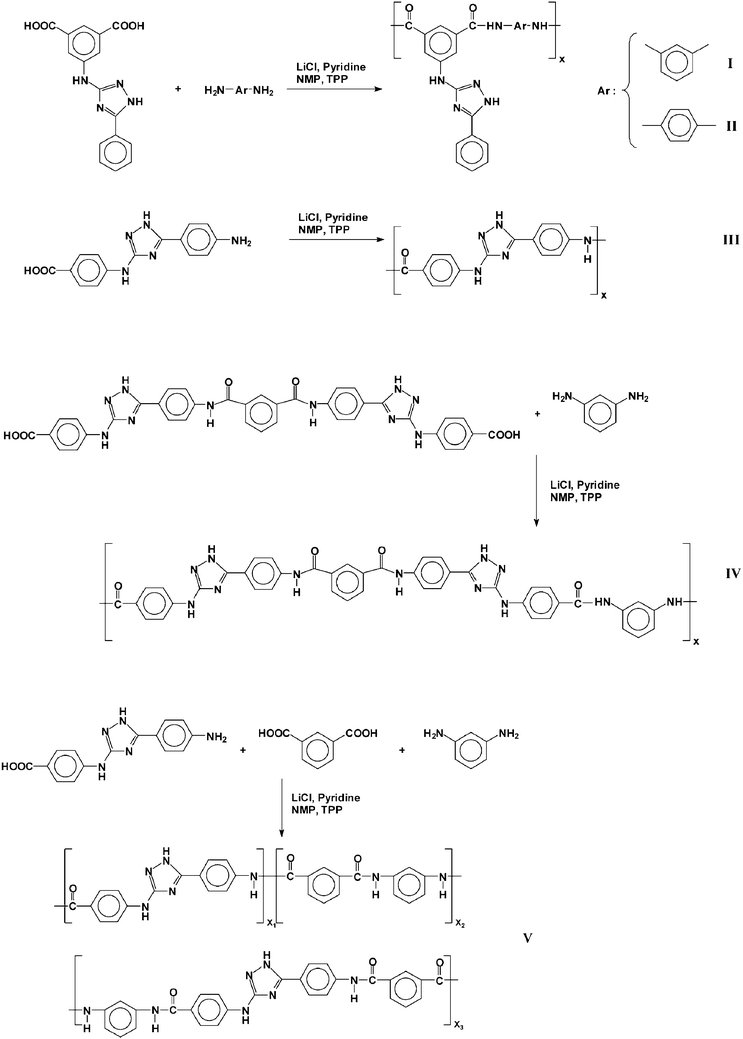 | ||
| Scheme 2 Polymer synthesis and associated codes. | ||
The following describes a typical polymerization reaction. In a 50 mL three-necked flask fitted with mechanical stirring, 10 mmol of diamine and 10 mmol of diacid (polymers I, II and IV), or 10 mmol of the amino acid (A-B monomer, polymer III), or 5 mmol of the amino acid, 5 mmol of MPD, and 5 mmol of isophthalic acid (copolymer V) and lithium chloride (1.4 g polymers I, II, and IV; 2.6 for polymer III and 2.0 g for copolymer V) were dissolved in a mixture of 6 mL of pyridine, 22 mmol of TPP and 20 mL of NMP (28 mL of NMP for copolymer III). The solution was stirred and heated at 110 °C under a dry nitrogen blanket for 4 h. The system was then cooled to room temperature and the solution was precipitated in 300 mL of methanol to render a swollen, fibrous precipitate. The polymer obtained was filtered and washed with methanol and distilled water, after which it was extracted with acetone for 24 h in a Soxhlet, and dried in a vacuum oven at 120 °C overnight. The yields were quantitative for all of the synthesised polymers.
Differential scanning calorimetry (DSC) data were recorded on a Perkin–Elmer Pyris I analyser with 10 mg of sample under a nitrogen atmosphere at a scan rate of 20 °C min−1. Thermogravimetric analysis (TGA) data was recorded on a 5 mg sample under a nitrogen or oxygen atmosphere on a TA Instrumenta Q50 TGA analyser at a scan rate of 10 °C min−1.
Polymer solubility was determined by mixing 10 mg of polymer with 1 mL of solvent, followed by stirring for 24 h at room temperature.
Polymer films were prepared by evaporation of cast solutions in NMP or DMA. Solutions of 8% by polymer weigh for polyamide I in NMP/DMA (50%, v/v) and 7% for polymer II and V in DMA were used. The solvent was eliminated by heating at 100 °C for 4 h in an air-circulating oven, and then at 120 °C for 4 h under vacuum (1 mm Hg). To determine the tensile properties of the polymers, strips (5 mm in width and 30 mm in length) were cut from polymer films of 30–45 μm thickness on a Hounsfield H10KM Universal Testing Dynamometer at 20 °C. Mechanical clamps were used and an extension rate of 5 mm min−1 was applied using a gauge length of 10 mm. At least six samples were tested for each polymer and the data was then averaged.
Water absorption measurements were determined gravimetrically at room temperature. Powdered polymeric samples of about 300 mg, previously dried at 120 °C for 24 over phosphorus pentoxide, were placed in a closed box containing a saturated aqueous solution of NaNO2 at 20 °C, which provided a relative humidity of 65%. The samples were periodically weighed over a period of 24 h and then allowed to remain in contact with this atmosphere for a further 8 days until they had equilibrated and presented no further changes in weight.
The solid-liquid extraction of CrIII, FeIII, CoII, NiII, CuII, ZnII CdII, HgII and PbII as their nitrate or chloride salts was carried out as follows. Approximately 10 mg of the appropriate polymer was shaken with 1 mL of an aqueous solution of the metal salt for a week at 25 °C (from previous kinetic experiments on extractions, equilibrium could be assumed in less than three days). The molar ratio of each cation to the triazole heterocycle subunits was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After filtration, the concentration of each cation in the liquid phase was determined by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500). Consecutive dilutions of sample aliquots with ultra-pure water/nitric acid (5% v/v) were performed to reach concentrations in the range of the calibration curve (from 0 to 40 ppb) and direct information regarding the extraction percentages of metal ions by polyamides was obtained.
:1. After filtration, the concentration of each cation in the liquid phase was determined by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500). Consecutive dilutions of sample aliquots with ultra-pure water/nitric acid (5% v/v) were performed to reach concentrations in the range of the calibration curve (from 0 to 40 ppb) and direct information regarding the extraction percentages of metal ions by polyamides was obtained.
Results and discussion
This work describes the properties and synthesis of aromatic polyamides and copolyamides bearing 1,2,4-triazole moieties linked to amine groups in the main and pendent chains. The triazole is a five-member cycle containing three nitrogens, which open the possibility to interchain interactions of this cycle with the polar amide and amine groups, thus deeply influencing the polymer properties and characteristics.Monomer and polymer synthesis and characterisation
The reaction steps carried out to obtain the diacid monomers (2, 5) and the amino acid A-B monomer (4) were easy and well documented from an organic chemistry viewpoint, and produced the monomers from widely available and cheap chemicals in good yield and purity. The preparation procedure of intermediates 1 and 3, and the reaction mechanism, were previously described by Kodomari et al.20 Purification by crystallization or solution-reprecipitation methods yielded monomers of high purity, as demonstrated by NMR spectra. The intermediates and monomers were characterised by IR, 1H and 13C NMR spectroscopy, thereby fully confirming the chemical structure of all the products.The polyamides have been synthesised by combining m-phenylenediamine or p-phenylenediamine with the diacid monomers 2 and 5, by homopolymerising the A-B monomer 4, or by copolymerizing the A-B monomer 4 with isophthalic acid and m-phenylenediamine. The polymers were synthesised according to the method described by Yamazaki et al. and others.21 Their structures are shown in Scheme 2, and the inherent viscosities are given in Table 1. As an illustrative example, Fig. 1 shows the chemical characterisation of polymer I. The inherent viscosity data, which was higher than 0.50 dL/g, and the 1H and 13C spectra were consistent with a high degree of polycondensation, and the peaks were assigned on the basis of model compounds, the ab initio theoretical analysis (discussed below) and the monomer's spectra.
| Polymer | ηinh (dLg−1) | Solubilitya | |||||
|---|---|---|---|---|---|---|---|
| DMSO, DMA | DMF | NMP | m-cresol | THF, CHCl3, CH, AcOH | Oleum | ||
| a CH = cyclohexanone; ++ = Soluble at room temperature; + = soluble on heating; +− = partially soluble; − = insoluble. b Inherent viscosity in oleum. c Inherent viscosity in a solution of LiCl in NMP (0.1g mL−1). | |||||||
| I | 0.58 | ++ | ++ | ++ | +− | — | ++ |
| II | 1.04 | ++ | ++ | + | — | — | ++ |
| III | 0.38 b | − | − | − | − | − | ++ |
| 0.68 c | |||||||
| IV | 0.50 | ++ | + | ++ | — | — | ++ |
| V | 0.56 | ++ | +− | ++ | — | — | ++ |
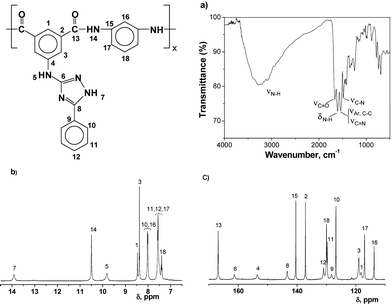
The 1,2,4-triazole heterocycle showed the tautomerism equilibrium depicted in Scheme 3, and exhibited acid/base behaviour consistent with their pKas.22 The presence of the tautomers in the polymer solutions was detected by 1H NMR experiments, although the spectrum of polyamide I depicted in Fig. 1 was obtained with selected experimental conditions in order to get a spectral pattern without the splitting of the triazole protons arising from the tautomerism of the heterocycle.
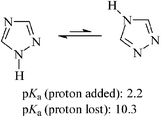 | ||
| Scheme 3 Tautomerism and acidity of 1,2,4-triazoles. | ||
The tautomerism equilibriums in the polyamides were studied by 1H NMR, as depicted in Fig. 2. The results showed that at 20 °C and at relatively concentrated samples, two different sharp signals were observed for both the amino and triazole heterocycle protons in the 1H NMR spectrum of polymer I. The increase in the experiment temperature from 20 °C to 100 °C caused a significant broadening of the proton signals of the triazole cycle and a collapse of the three signals of the amine proton. We believe that these results confirmed the tautomerism equilibrium, as at higher temperatures, the faster chemical proton interchange gave rise to a broadening in the signals of the cycle protons. Thus, the NMR pattern showed the behaviour of a unique overage cycle, due to the NMR time scale, thus giving rise to a relatively sharp unique signal of the amine proton. The broadening of these proton signals by decreasing the concentration of the 1H NMR samples has also been detected, as depicted in Fig. 2.
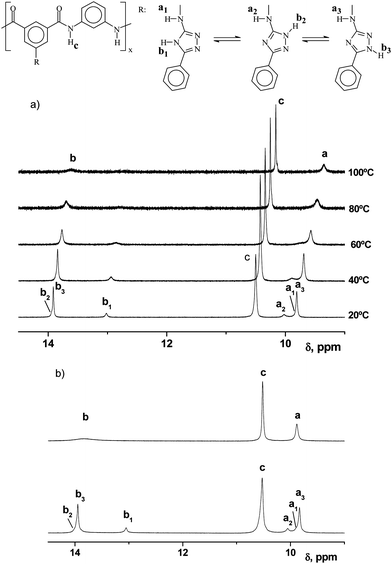 | ||
| Fig. 2 (a) Temperature dependence of the 1H NMR spectra of polyamide I. (b) Concentration dependence of the 1H NMR spectra of polyamide I: (upper) diluted conditions, 5.6 × 10−3 mol of structural unit/L; (lower) concentrated conditions, 8.4 × 10−2 mol of structural unit/L. | ||
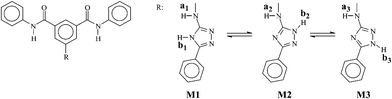
The tautomerism was also studied theoretically (ab initio) by gas-phase density functional theory (DFT) calculations.23 The estimated energies and 1H NMR chemical shifts of the tautomers of the 1,2,4-triazole moiety of a polyisophthalamide model compound are shown in Table 2. The relative energy of the tautomers and the predicted chemical shifts were used to assign the NMR signals of the synthesised polymers.
|
|
|||
|---|---|---|---|
| Model compound | Energy (relative energy)a/kJ mol−1 | Estimated 1H NMR chemical shiftsa | |
| Ha | Hb | ||
| a : The geometry optimisation and energy evaluations were performed with Spartan software using density functional theory (DFT) calculations at the B3LYP/6-31G* level of theory. | |||
| M1 | −4093735.74 (40.0) | 5.66 (a1) | 8.05 (b1) |
| M2 | −4093766.31 (9.44) | 5.73 (a2) | 8.79 (b2) |
| M3 | −4093775.75 (0.0) | 5.54 (a3) | 9.86 (b3) |
Properties of the polymers
| Polymer | DSC | TGA | ||||||
|---|---|---|---|---|---|---|---|---|
| T g/°C | N2 atmosphere | O2 atmosphere | ||||||
| T 5 /°C | T 10 /°C | Char yield (%) | T 5 /°C | T 10 /°C | Char yieldc (%) | LOI | ||
| a 5% weight loss. b 10% weight loss. c Limiting oxygen index. | ||||||||
| I | 293 | 380 | 400 | 50 | 380 | 400 | — | 37.5 |
| II | 306 | 390 | 405 | 46 | 385 | 405 | — | 35.9 |
| III | 375 | 400 | 420 | 50 | 390 | 415 | 3 | 37.5 |
| IV | 345 | 385 | 415 | 48 | 365 | 400 | — | 36.7 |
| V | 295 | 380 | 415 | 53 | 380 | 410 | 3 | 38.7 |
Polyamides I and II showed strong similarity with a related polyamide in terms of chain stiffness, lateral volume and polarity of the bulky side group, as poly(m-phenylene 5-benzoylaminoisophthalamide) and poly(p-phenylene 5-benzoylaminoisophthalamide), which have a Tgs of 298 and 318 °C, respectively.10
Conversely, the Tg of polymer III was as high as 375 °C, showing the influence of the heterocycle in the structural unit, the rigidity of the backbone imposed by the rigid A-B monomer and the extra hydrogen bond interactions, as compared with conventional aramids with amine/amide, amide/heterocycle, heterocycle/heterocycle or amine/heterocycle subgroups.
The Tgs of polymer IV and copolymer V were closely related to the transition values of polymer III (375 °C) and poly(m-phenylene isophthalamide) (275 °C).
The thermogravimetric data of polymers, summarised in Table 3, confirmed their good thermal stability. The temperature of 10% weight loss of these polyamides under nitrogen and oxygen atmospheres was higher than 400 °C in all cases. TGA thermograms for polyamide II, depicted in Fig. 3, show a similar behaviour of the polyamides in both oxygen and nitrogen atmospheres, and had very similar decomposition temperatures. This was an important observation because most polymer applications occur under normal atmospheric environments.
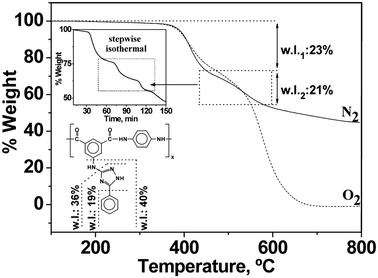 | ||
| Fig. 3 TGA of polyamide II under nitrogen and oxygen atmospheres (w.l.: weight loss percentage). The auto stepwise isothermal technique was carried out under nitrogen atmosphere. | ||
Up to three degradation steps were detected, but most of the polymers showed only two. As an illustrative example, Fig. 3 depicts the TGA degradation pattern and the data interpretation of polymer II. When the second decomposition step was completed, the loss of the complete pendant group was observed. In order to improve weight change resolution, an auto stepwise isothermal experiment was performed for polyamide II (the use of the auto stepwise mode gives the highest possible degree of resolution between successive decomposition events, such as those associated with complex degradation processes). In this type of experiment the sample was heated at a constant rate until a weight loss was observed, continuing with the isothermal conditions until the decomposition event took place, and then resuming heating until the next weight loss. Using this approach, overlapping decomposition processes could sometimes be separated.
The char yield at a given temperature was intimately related with the thermal behaviour of the polymers. Thus, the char yield of these polymers in a nitrogen atmosphere was significant, being higher than 48% at 800 °C. The char yield could be used to estimate the limiting oxygen index (LOI) of the polymers using the experimental Van Krevelen equation (LOI = 17.5 + 0.4 CR, where CR is the char yield in % weight).24 The polymers had LOI values higher than 36, meaning that our polymers could be classified as self-extinguishing polymers (LOI is defined as the minimum fraction of oxygen in a mixture of oxygen and nitrogen that will support combustion after ignition). As air contains approximately 21% oxygen, any material with a LOI lower than 21 will probably not burn in an open air situation.
Polyamide tensile strength and the Young's modulus (Table 4) were found to be around 40–44 MPa and 1.3–1.7 GPa, respectively. The mechanical properties of these polyamides could be considered reasonably good for non-oriented films made by casting on a laboratory scale with no post-thermal treatment.14,25
| Polymer | Mechanical properties | Moisture absorption | ||||
|---|---|---|---|---|---|---|
| Tensile strength/MPa | Young's Modulus/GPa | Elongation (%) | Water uptake (%) | Molecules of H2O/repeating unit | Molecules H2O/equiv of amino-triazole a | |
| a Considering an uptake of 0.6 water molecules per amide group. b Dry film. c Film stored in a 65% relative humidity atmosphere. d film immersed in pure water for 24 h. | ||||||
| I | —b | — | — | 13.3 | 2.9 | 1.7 |
| 37 c | 1.5 | 2.4 | ||||
| 43 d | 1.2 | 3.8 | ||||
| II | 44 b | 1.7 | 4.4 | 13.3 | 2.9 | 1.7 |
| 50 c | 1.5 | 10 | ||||
| 55 d | 1.4 | 18 | ||||
| III | — | — | — | 12.3 | 1.9 | 1.3 |
| IV | — | — | — | 12.0 | 5.5 | 1.5 |
| V | 40 bcd | 1.4 | 2.2 | 10.3 | 3.0 | 1.2 |
The influence of water uptake on mechanical properties is shown in Table 4. Increasing levels of water absorption were found to reduce the Young's modulus and enhance the tensile strength and elongation in polyamide I and II, thus enhancing their tenacity. These results agreed with our previous experiences in film handling. Dry strips of polyamide I were fragile and could be transformed into a creasable material after storing in a 65% relative humidity (r.h.) atmosphere or after immersing in pure water. The storage at 65% r.h. or immersion in water of polyamide II enhanced its tensile strength by 13% and 23%, respectively, while the Young's modulus fell by 18% and the elongation increased by 310% when the film was immersed in water. Conversely, the mechanical behaviour of polyamide V remained nearly constant with increasing quantities of water uptake.
Strips of polyamide II, previously immersed in pure water until the water uptake equilibrium was achieved, were also repeated subjected to tensile tests before fracture. The chain orientation induced by mechanical stretching enhanced the tensile strength and Young's modulus by 66% and 50%, respectively.
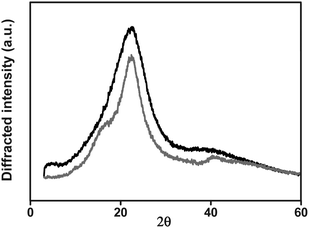 | ||
| Fig. 4 X-Ray diffraction patterns of polyamide I (black line) and III (grey line). | ||
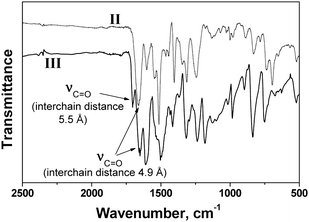 | ||
| Fig. 5 FT-IR spectra of polymers II and III. | ||
Conversely, the rigid and ordered polyamide structure derived from the polymerization of the amino acid monomer (polyamide III, monomer 4) exhibited an extremely low solubility, and could only be dissolved in concentrated sulfuric acid and in aprotic solvents with salts (LiCl) as solubility promoters. The polymer was not degraded by the sulfuric acid, as shown by the variation of the inherent viscosities of solutions of III in oleum (Table 5). The insolubility of this polymer compared well with the solubility of poly(p-phenylene terephthalamide).1 On the other hand, the copolymerisation of the amino acid monomer (4) with equimolecular quantities of isophthalic acid and m-phenylene diamine (Scheme 2) gave rise to a random copolymer (V) with a structure resembling both polyamide III and poly(m-phenylene isophthalamide), and which had an enhanced solubility compared with III.
The polyamides described herein had from two (I, II, III) to four (IV) amide groups, and from one (I, II, III) to two (IV) triazole and amine moieties per structural unit, with the resulting copolymers having complex structures. As it has been previously outlined, the amide, the amine and the triazole cycle moieties are highly polar groups, which influenced the water uptake of the polymers through hydrogen bonding interactions with water.
The measurement of the isothermal absorption of water at 65% r.h. gave values that correlated to the polyamide chemical structure. Table 4 shows the water absorption percentages for polyamides, the molecules of water per structural unit, and estimated molecules of water per amine-triazole moiety. The latter was calculated on the basis of an uptake of 0.6 water molecules per amide group, predicted from the water absorbed by poly(m-phenylene 5-benzoylaminoisophthalamide).16
The polyamide water absorption rates were high, ranging between 10.3–13.3%, with the higher values corresponding to the polymers with pendant structures, probably due to the amorphous structure of the polyamides and also because of the higher interchain distances imposed by the bulkiness of the lateral groups.
Regarding the number of water molecules per repeating unit, a range between 1.9 and 5.5 was observed, with the higher values corresponding to the polymer with the higher content of polar moieties in the polymeric structural unit (IV). The remarkably low value corresponding to polymer III could be attributed to the order derived from the linear and regular polymer structure that gave rise to crystallinity and a higher chain packing.
The estimated water uptake per triazole-amine group varied between 1.2 and 1.7 molecules, which gave an idea of the hydrophilic character of these two groups and of the influence they have on the polyamides.
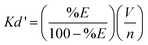 | (1) |
The solid-liquid extraction selectivity, αS,L, is the ratio of two distribution coefficients (eqn (2)). The HgII cation was taken as the reference, as it has the highest distribution coefficient (Kd′M2).
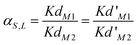 | (2) |
Table 6 depicts the solid-liquid extraction results for the elimination of several cations from aqueous solution using polyamide I as the extractant. We chose this polyamide because of its high water uptake as well as its all-meta phenylene polymer backbone conducting to lower linearity and less rigid macromolecular structure. Extraction levels between 25 and 36% were found for tested cations, except for HgII, which had an extraction level from aqueous solution close to 100%.
| Salt | % E | Kd′/L mol−1 | α × 103 (Kdi′/KdHg′) |
|---|---|---|---|
| Cr(NO3)3 | 36 | 21 | 8 |
| Fe(NO3)3 | 30 | 16 | 6 |
| Co(NO3)2 | 35 | 21 | 8 |
| Ni(NO3)2 | 35 | 21 | 8 |
| Cu(NO3)2 | 36 | 21 | 8 |
| ZnCl2 | 25 | 14 | 5 |
| Cd(NO3)2 | 34 | 20 | 8 |
| Hg(NO3)2 | 99 | 27 × 102 | 1 × 103 |
| Pb(NO3)2 | 31 | 16 | 6 |
The extraction level of mercury cations from aqueous media by solid polyamide phases points to future applications in the fields of water decontamination, membrane preparation for selective cation transport and in sensing materials, among other applications.
Conclusion
This work describes the synthesis and characterization of five new aromatic polyamides bearing the triazole heterocycle in the pendant or main chain structures. These polyamides showed a good thermal behaviour and good solubility that makes them suitable for solution transformation. These properties, in turn, enable tough colourless transparent films to be cast, which have properties which unusually enhance with increasing water content in the material. The polar triazole and the amide groups greatly increased the hydrophilicity of the polyamides, making these polymers promising materials for the production of water treatment or gas separation membranes. Moreover, the triazole group was found to be a chelating, or host, unit for toxic heavy metal cations, thus making these polyamides effective solid extractant phases in the extraction and elimination of mercury cations from water media (elimination of nearly 100% of the HgII cations was observed in the experimental conditions).Acknowledgements
The authors gratefully acknowledge the financial support provided by the Spanish Ministerio de Educación y Ciencia – Feder (MAT2008-00946/MAT) and by the Caja de Burgos.References
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef CAS.
- M. Trigo-López, P. Estévez, N. San-José, A. Gómez-Valdemoro, F. C. García, F. Serna, J. L. de la Peña and J. M. García, Recent Pat. Mater. Sci., 2010, 2, 190–208 Search PubMed.
- (a) P. E. Cassidy, Thermally Stable Polymers, Dekker, New York, 1980 Search PubMed; H.H. Yang, Aromatic High-Strength Fibers. Wiley, New York, 1989 Search PubMed; (b) J. K. Fink, High Performance Polymers, William Andrew Inc, New York, 2008 Search PubMed; L. Vollbracht, Aromatic Polyamides, in Comprehensive Polymer Science, ed. G. Allen, B. Bevington, G. V. Eastmond, A. Ledwith, S. Russo and P. Sigwald, Pergamon Press, Oxford, 1989, vol. 5, ch. 22, pp. 373–383 Search PubMed; (c) J. Gallini, Polyamides, aromatic, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, New York, 2005, vol. 3, pp. 558-84 Search PubMed; (d) S. Ozawa and K. Matsuda, Aramid copolymer fibers, in Handbook of Fiber Science and Technology. Vol III: High Technology Fibers: Part B, ed. M. Lewin and J. Preston, Marcel Dekker Inc., New York, 1989, vol. 3, ch. 1, pp. 1–34 Search PubMed; (e) D. Tanner, J. A. Fitzgerald, P. G. Riewald and W. F. Knoff, Aramid structure/property relationships and their role in applications development, in Handbook of Fiber Science and Technology. Vol III: High Technology Fibers: Part B, ed. M. Lewin and J. Preston, Marcel Dekker Inc., New York, 1989, vol. 3, ch. 2, pp. 35–80 Search PubMed; (f) H. H. Yang, Nomex aramid fiber, in Handbook of Fiber Science and Technology. Vol III: High Technology Fibers: Part C, ed. M. Lewin and J. Preston, Marcel Dekker Inc., New York, 1993, vol. 3, ch. 2, pp. 77–178 Search PubMed; (g) J. Peterson, Aromatic Polyamides, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, John Wiley & Sons, Inc., New York, 1988, vol. 11, pp. 381–409 Search PubMed.
- (a) A. Abdolmaleki, Polym. Degrad. Stab., 2007, 92, 292–928 CrossRef CAS; (b) I. Sava, M. D. Iosip, M. Bruma, C. Hamciuc, J. Robison, L. Okrasa and T. Pakula, Eur. Polym. J., 2003, 39, 725–738 CrossRef CAS.
- (a) K. Faghihi and Z. Mozaffari, J. Appl. Polym. Sci., 2008, 108, 1152–1157 CrossRef CAS; I. In and S. Y. Kim, Polymer, 2006, 47, 547–552 CrossRef CAS; (b) B. Tamami and H. Yeganeh, Polymer, 2001, 42, 415–420 CrossRef CAS; (c) S. Mehdipour-Ataei and H. Heidari, Macromol. Symp., 2003, 193, 159–167 CrossRef CAS.
- F. A. Bottino, G. Di Pasquale, L. Scalia and A. Pollicino, Polymer, 2001, 42, 3323–3332 CrossRef CAS.
- (a) J. Tan, C. Wang, W. Peng, G. Li and J. M. Jiang, Polym. Bull., 2009, 62, 195–207 CrossRef CAS; (b) L. Cheng and G. Jian, J. Appl. Polym. Sci., 2004, 92, 1516–1520 CrossRef CAS.
- (a) R. R. Pal, P. S. Patil, M. M. Salunkhe, N. N. Maldar and P. P. Wadgaonkar, Polym. Int., 2005, 54, 569–575 CrossRef CAS; (b) A. D. Sagar, R. D. Shingte, P. P. Wadgaonkar and M. M. Salunkhe, Eur. Polym. J., 2001, 37, 1493–1498 CrossRef CAS; (c) H. S. Patel, R. R. Shah and K. C. Patel, Int. J. Polym. Mater., 2007, 56, 499–510 CrossRef CAS.
- (a) S. H. Hsiao, C. P. Yang, C. W. Chen and G. S. Liou, Eur. Polym. J., 2005, 41, 511–517 CrossRef CAS; (b) S. Bhuvana, M. Madhumathi and M. Sarojadevi, Polym. Bull., 2006, 57, 61–72 CrossRef CAS; (c) K. I. Aly, M. H. Wafadan and M. A. Hussein, J. Appl. Polym. Sci., 2009, 112, 513–523 CrossRef CAS.
- A. Banihashemi and H. Firoozifar, Eur. Polym. J., 2003, 39, 281–289 CrossRef CAS.
- S. H. Hsiao, C. W. Chen and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3302–3313 CrossRef CAS.
- (a) M. Ghaemy, H. Behmadi and R. Alizadeh, Chin. Chem. Lett., 2009, 20, 961–964 CrossRef CAS; (b) V. Ayala, E. M. Maya, J. M. García, J. G. de la Campa, A. E. Lozano and J. de Abajo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 112–121 CrossRef CAS.
- J. H. Kim, Y. H. Kim, Y. J. Kim, J. C. Won and K. Y. Choi, J. Appl. Polym. Sci., 2004, 92, 178–185 CrossRef CAS.
- J. J. Ferreiro, J. G. de La Campa, A. E. Lozano and J. de Abajo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5300–5311 CrossRef CAS.
- (a) S. Elshani, P. Apgar, S. Wang and C. M. Wai, J. Heterocycl. Chem., 1994, 31, 1271–1274 CrossRef CAS; (b) L. Uzun, A. kara, B. Osman, E. Yilmaz, N. Besirli and A. Denizli, J. Appl. Polym. Sci., 2009, 114, 2246–2253 CrossRef CAS; (c) A. Kumar and P. S. Pandey, Tetrahedron Lett., 2009, 50, 5842–5845 CrossRef CAS; (d) J. Zhan, D. Tian and H. Li, New J. Chem., 2009, 33, 725–728 RSC.
- H. C. Hung, C. W. Cheng, I. T. Ho and W. S. Chung, Tetrahedron Lett., 2009, 50, 302–305 CrossRef CAS.
- (a) B. Liu, G. C. Guo and J. S. Huang, J. Solid State Chem., 2006, 179, 3136–3144 CrossRef CAS; (b) G. Mahmoudi, A. Morsali and L. G. Zhu, Z. Anorg. Allg. Chem., 2007, 633, 539–541 CrossRef CAS; (c) K. C. Chang, I. H. Su, A. Senthilvelan and W. S. Chung, Org. Lett., 2007, 9, 3363–3366 CrossRef CAS.
- V. Calderón, F. C. García, J. L. de la Peña, E. M. Maya and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2270 CrossRef CAS.
- (a) V. Calderón, F. García, J. L. de la Peña, E. V. Maya, A. E. Lozano, J. I. de la Campa, J. de Abajo and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4063–4075 CrossRef CAS; (b) V. Calderón, G. Schwarz, F. García, M. J. Tapia, A. J. M. Valente, H. D. Burrows and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6252–6269 CrossRef CAS; (c) J. M. García, F. C. García and F. Serna, J. Polym. Res., 2007, 14, 341–350 Search PubMed; (d) F. Serna, F. García, J. L. de la Peña and J. M. García, High Perform. Polym., 2008, 20, 19–37 CAS; (e) N. San-José, A. Gómez-Valdemoro, F. C. García, F. Serna and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4026–4036 CrossRef CAS; (f) N. San-José, A. Gómez-Valdemoro, F. C. García, J. L. de la Peña, F. Serna and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5398–5407 CrossRef CAS; (g) M. J. Tapia, A. J. M. Valente, H. D. Burrows, V. Calderón, F. García and J. M. García, Eur. Polym. J., 2007, 43, 3838–3848 CrossRef CAS; (h) V. Calderón, F. Sena, F. García, J. L. de la Peña and J. M. García, J. Appl. Polym. Sci., 2007, 106, 2875–2884 CrossRef CAS; (i) N. San-Jose, A. Gómez-Valdemoro, F. Clemente García, V. Calderón and J. M. García, React. Funct. Polym., 2008, 68, 1337–1345 CrossRef CAS; (j) N. San-José, A. Gómez-Valdemoro, P. Estevez, F. C. García, F. Serna and J. M. García, Eur. Polym. J., 2008, 44, 3578–3587 CrossRef CAS; (k) A. Gómez-Valdemoro, V. Calderón, N. San-José, F. C. García, J. L. de la Peña and J. M. García, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 670–681 CrossRef CAS; (l) N. San-José, A. Gómez-Valdemoro, V. Calderón, J. L. de la Peña, F. Serna, F. C. García and J. M. García, Supramol. Chem., 2009, 21, 337–343 CrossRef CAS.
- M. Kodomari, M. Suzuki, K. Tanigawa and T. Aoyama, Tetrahedon Lett., 46, pp. 5841–5843 Search PubMed.
- (a) N. Yamazaki, F. Higashi and J. Kawataba, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 2149–2154 CrossRef CAS; (b) D. J. Liaw, B. Y. Liaw and M. Q. Jeng, Polymer, 1998, 39, 1597–1607 CrossRef CAS; (c) D. J. Liaw, B. Y. Liaw and C. Y. Chung, Acta Polym., 1999, 50, 135–140 CrossRef CAS; (d) D. J. Liaw and B. Y. Liaw, Macromol. Chem. Phys., 1999, 200, 1326–1332 CrossRef CAS; (e) D. J. Liaw, B. Y. Liaw, J. R. Chen and C. M. Yang, Macromolecules, 1999, 32, 6860–6863 CrossRef CAS; (f) D. J. Liaw, B. Y. Liaw and C. M. Yang, Macromolecules, 1999, 32, 7248–7250 CrossRef CAS; (g) D. J. Liaw, P. N. Hsu, W. H. Chen and S. L. Lin, Macromolecules, 2002, 35, 4669–4676 CrossRef CAS; (h) D. J. Liaw, F. C. Chang, M. K. Leung, M. Y. Chou and K. Muellen, Macromolecules, 2005, 38, 4024–4029 CrossRef CAS; (i) D. J. Liaw, C. C. Huang and W. H. Chen, Polymer, 2006, 47, 2337–2348 CrossRef CAS.
- J. A. Joule and K. Mills, Heterocycle Chemistry, Blackwell Science, Oxford, 2000 Search PubMed.
- Spartan'08. Wavefunctin, Inc. Irving, CA; Spartan'08 for Windows, Macintosh and Linux. Tutorial and User's Guide. Wavefunctin, Inc. Irving, 2008 Search PubMed.
- D. W. Van Krevelen and K. te Nijenhuis, Properties of Polymers. Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions, Elsevier, Amsterdam, 4th edn, 2009, pp. 855–57 Search PubMed.
- (a) A. Marcos-Fernández, A. E. Lozano, J. de Abajo and J. G. de la Campa, Polymer, 2001, 42, 7933–7941 CrossRef CAS; (b) E. Ferrero, J. F. Espeso, J. G. de la Campa, J. de Abajo and A. E. Lozano, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3711 CrossRef CAS.
- (a) B. Dunjic, A. Favre-Réguillon, O. Duclaux and M. Lemaire, Adv. Mater., 1994, 6, 484–486 CrossRef CAS; (b) A. Duhart, J. F. Dozol, H. Rouquette and A. Deratani, J. Membr. Sci., 2001, 185, 145–155 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |