Polymer–protein conjugates: an enzymatic activity perspective
Marc A.
Gauthier
*a and
Harm-Anton
Klok
b
aSwiss Federal Institute of Technology Zürich (ETHZ), Drug Formulation & Delivery, Institute of Pharmaceutical Sciences, Wolfgang-Pauli Str. 10, HCl J 396.4, CH-8093, Zürich, Switzerland. E-mail: marc.gauthier@pharma.ethz.ch; Fax: +41 44 633 1314; Tel: +41 44 633 7329
bÉcole Polytechnique Fédérale de Lausanne (EPFL), Institut des Matériaux and Institut des Sciences et Ingénierie Chimiques, Laboratoire des Polymères, Bâtiment MXD, Station 12, CH-1015, Lausanne, Switzerland
First published on 11th June 2010
Abstract
Proteins have been modified with polymers in diverse manners over the past 30 years. However, while proteins have been used to prepare many functional constructs, they are sensitive biomolecules and their bioactivity can be either positively or negatively influenced by many different aspects of polymer modification. The primary focus of this review article is to highlight the opportunities offered by new trends in protein modification, and specifically how they influence the overall biological activity of the conjugate, including its dependence on temperature and pH. We survey the effect of polymer molecular weight, number of conjugated polymer chains, polymer coupling strategy (including random versus site-specific coupling, “grafting from”, and multi-point covalent attachment), polymer architecture (including branched and comb-type), polymer interactions with the protein (including electrostatic and host–guest interactions), polymer interactions with enzyme substrate, and polymer biodegradability. We have selected six enzymes, which have been extensively modified with polymers in diverse fashions in the literature, as basis for this discussion. These proteins are L-asparaginase, alpha-chymotrypsin, trypsin, lysozyme, bovine serum albumin, and papain. This review includes polymers such as poly(ethylene glycol) (PEG), polysaccharides, polypeptides, and other synthetic (vinyl) polymers. From the discussed literature we attempt to extract tentative general trends observed between state-of-the-art methods of preparing protein–polymer conjugates and the activity of the conjugate.
 Marc A. Gauthier | Marc A. Gauthier was awarded his Ph.D. in 2007 for his work with T.H. Ellis and X.X. Zhu at the University of Montreal (Montreal, Canada). During this period he received fellowships from the Natural Science and Engineering Council of Canada and the Fonds Québecois de la Recherche sur la Nature et les Technologies (FQRNT). He then pursued post-doctoral research (FQRNT) with H.-A. Klok (EPFL, Switzerland) in the field of peptide/protein–polymer conjugation. He currently holds the position of Research Fellow at the Institute of Pharmaceutical Sciences (IPW) at the Swiss Federal Institute of Technology Zürich (ETHZ) (Zürich, Switzerland), where he is pursuing independent research involving therapeutic protein conjugates. |
 Harm-Anton Klok | Harm-Anton Klok is Full Professor at the Institutes of Materials and Chemical Sciences and Engineering at the Ecole Polytechnique Fédérale de Lausanne (EPFL) (Lausanne, Switzerland). His current research interests include peptide/protein-based materials and peptide/protein-polymer hybrids, surface-initiated polymerization and polymer brushes, controlled/“living” polymerization and macromolecular engineering as well as dendritic and hyperbranched polymers. Harm-Anton Klok is recipient of the 2007 Arthur K. Doolittle Award of the American Chemical Society and is Associate Editor of the American Chemical Society journal Biomacromolecules. |
1. Introduction
Background
Over the past 30 years proteins have been extensively modified with either well- or ill-defined polymers by means of either well- or ill-defined coupling chemistry. By far, the desire to overcome the intrinsic limitations of protein therapeutic agents has been the driving force for the developments in this field, given that approximately 25% of all new medicines approved by the American Food and Drug Administration are protein-based.1 Initially, the philosophy for overcoming the immunogenicity of therapeutic proteins, their renal elimination, and their proteolysis while maintaining therapeutic activity has been to modulate the number, the length, and to a certain extent the architecture (i.e., linear versus branched) of the polymer segments used for preparing the conjugate. Early on, many natural polymers as well as synthetic polymers such as poly(ethylene glycol) (PEG) have been evaluated for this purpose. Possibly due to the fact that PEG was one of the first non-immunogenic, well-defined (hetero)telechelic polymers capable of conveying “stealth” properties to proteins, it is not surprising that the PEGylation (i.e., the covalent attachment of PEG chains) of therapeutic proteins has dominated this field for many years2,3 and that several therapeutic PEGylated proteins have reached the market.4–6 More recently, the philosophy for overcoming the shortcomings of therapeutic proteins with minimal detrimental effect on protein biological activity has changed to controlling the site at which the polymer is attached to the protein.7–9 This has become a possibility with the development of a veritable (bio)chemical toolbox of orthogonal coupling strategies for residue- or site-specific protein modification.10–22 Concurrently, access to well-defined (hetero)telechelic polymers other than PEG has become possible due to advances in the field of controlled/“living” polymerization.23–27 These combined developments have lead to a boom in the number of studies implicating protein–polymer conjugates, whether for therapeutic purposes or not. With these advances, many new possibilities are available for adjusting the properties of the final conjugate. One aspect which is of prime importance in the field is that while proteins are used to prepare many functional constructs, they are sensitive biomolecules and their bioactivity can be either positively or negatively influenced by many different aspects of polymer modification. The influence of polymer modification of proteins has previously been discussed by others in the context of PEGylation.3–6,28,29 However, a discussion which puts these findings into a broader context and which includes modern design and synthetic strategies, inaccessible to classic PEGylation, is lacking. The primary focus of this review article is to highlight the trends observed for these new developments in protein modification, and specifically how they influence the overall biological activity of the conjugate, including its dependence on temperature and pH. This contribution is divided into 6 sections which successively discuss the effect of the number and molecular weight of coupled polymer chains (i.e., lessons learned from PEGylation), polymer coupling strategy, polymer architecture, polymer interaction with the protein, polymer interaction with enzymatic substrate (vide infra), and polymer biodegradability.Scope
In order to have a reliable, comparable parameter with which to compare the biological properties of protein–polymer conjugates prepared over the past three decades, we have selected enzymes with well-defined and/or commercially available enzymatic substrates for discussion. We have avoided discussing proteins whose activity assays involve live cells, as these tests have more potential to be influenced by unrelated cell- or user-specific variables, which may potentially complicate or bias the establishment of trends within the literature. This implies that very recent and exciting research on conjugates prepared from interleukins, leptin, interferons, antibody fragments, streptavidin, or enzymes with poorly defined enzymatic assays fall outside the scope of this review. Polymer-modification of these types of proteins has been reviewed elsewhere.7,30,31 Given the vastness of the literature as well as the overwhelming number of publications in this field published every year, we have restricted our discussion to six enzymes. By no means is this contribution intended to be comprehensive and is rather geared towards providing some perspective in the properties of protein–polymer conjugates. The choice of these proteins was subjective but was made based on the criteria that they have been modified in numerous and diverse fashions by synthetic and/or natural polymers. This implies that not all selected proteins are necessarily of therapeutic interest and may simply be popular model proteins for proof-of-concept studies (e.g., owing to cost, availability, or structural considerations) or proteins useful for industrial applications, etc. We have also limited our discussion to water-soluble protein–polymer conjugates, thus eliminating the plethora of studies on enzymes immobilized on insoluble polymer matrices.The structural and enzymatic parameters of all discussed conjugates are listed in Tables 1–6 and serve as basis for the discussions herein. Enzymatic activity is always given as a percentage of that of the native enzyme and the numerical value is taken directly from the corresponding study, unless preceded by a “∼” which indicates that it was estimated from a figure. When only enzyme kinetic parameters such as kcat (turn over number) and KM (Michaelis constant) are given in the original manuscript, these values are used for discussion. From these parameters we make no attempt to calculate an enzymatic efficiency (kcat/KM) of the conjugates, as this parameter is ill-suited for comparing enzymes.32
2. Polymer molecular weight and number of chains
Over the past 30 years, proteins have been extensively modified with PEG in order to establish structure–activity relationships between molecular weight, coupling chemistry, and extent of modification on the activity of the final conjugate. PEGylation is an excellent tool for evaluating the effects of polymer molecular weight and degree of polymer coupling (to proteins) because it does not bear functional side-chains which could potentially interact with the protein and thus bias the results. Classically, the number and length of conjugated polymer chains is thought to correlate with the amount of polymer present on the surface of the enzyme, restricted diffusion of substrate towards the enzymes catalytic site, and reduced bioactivity. In this section we will treat the polymers as being chemically inert and verify the effect of these two parameters on enzyme activity. We will begin by discussing the lessons learned from the extensive history of PEGylation (as demonstrated by the multiple entries in Tables 1–6 involving PEG) and then we will discuss other polymers. This will be followed by a discussion of general trends which can be extracted from the analyzed literature, and which are summarized in Table 7.| Polymer | Residue (number)b | MW (chain, conjugate)c | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a ASNase is from E. coli unless otherwise mentioned. b Number of residues modified on protein. c Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscripts d (Mn) or e (Mv), the nature of the average molecular weight was not specified. f L-Asparagine. g Aspartic acid-β-p-nitroanilide. h Not given. i E.carotovora. | |||||||
| Synthetic polymers | |||||||
| MPEG | Lys (18–73) | 5 kDa | TCT | 6–40f, 15–50g | — | — | Effect of molecular weight, 33 |
| MPEG | Lys (77) | 750 Da | TCT | 12f, 20g | — | — | |
| MPEG | Lys (70) | 1.9 kDa | TCT | 14f, 25g | — | — | |
| MPEG | Lys (50) | 5 kDa | NHS | 30f | — | — | Ref. 36 |
| MPEG | Cys (4) | 5 kDa | bisAlk | 100 | — | — | Site-specific58 |
| MPEG | Cys (4) | 10 kDa | bisAlk | 100 | — | — | |
| MPEG | Cys (4) | 20 kDa | bisAlk | 100f | — | — | |
| MPEG2 | Lys (33–52) | 2 × 5 kDa | TCT | 11–30f | — | — | Branched polymer34 |
| MPEG2 | Lys(52) | 2 × 5 kDa | TCT | 8f | ∼KM,nat | — | Branched polymer91 |
| MPEG2 | Lys (2.5–30) | 2 × 5 kDa | TCT | 0–74h | — | — | Branched polymer35 |
| MPEG | Lys (35–49)i | 5 kDad | NHS | 110 | 3.33 × 10−3 | 11.83 | Effect of polymer architecture37,38 |
| MPEG2 | Lys (27–37)i | 2 × 5 kDad | NHS | 130–133f | 3.30 × 10−3 | 13 | |
| (3.31 × 10−3)f | (8.7)f | ||||||
| MPEG | Lys (73) | 5 kDa | TCT | 0.9 | — | — | Multi-site attachment. Comb-shaped74 |
| MPEG2 | Lys (52) | 2 × 5 kDa | TCT | 11 | — | — | |
| PM13 | Lys (46) | 13 kDa, n.c. | MA | 45.5 | — | — | |
| PM100 | Lys (31) | 100 kDa, n.c. | MA | 85.3f | — | — | |
| PMPEG5000-g-VP-co-MA) | Lys (30) | 77 kDa, n.c.d | MA | 58.6f | — | — | Multi-site attachment. Comb-shaped89 |
| P(VP-co-MA) | Lys (63) | 34 kDa, n.c.e | MA | 8.3f | — | — | Multi-site attachment90 |
| Polypeptides | |||||||
| P(DL-alanine) | Lys (23.5) | 5.4 kDad | NCA | 20 | 0.011 | — | Grafting from48 |
| P(DL-alanine) | Lys (17) | 2.2 kDad | NCA | 48 | — | — | |
| P(DL-alanine) | Lys (31.5)i | 3.5 kDad | NCA | 65f | 0.03 | — | |
| (0.011) | — | ||||||
| (0.04)i | — | ||||||
| Rat albumin | Lys (n.g.) | 69 kDa | GA | 60f | 2-3 × KM,nat | ∼12 chains/enzyme87 | |
| Silk fibroin | Lys (28–55) | 40–120 kDa, n.c. | GA | ∼40–80f | 0.84 (4.77) | — | Natural polymer72 |
| Sericin | Lys (45) | 20–60 kDa, n.c. | GA | ∼25f | 0.159 (10.4)f | — | Activity for pH = 7.493 |
| Carbohydrate polymers | |||||||
| N,O-CM-Chit | Lys (∼10)b | 40, 522 kDa | GA | ∼80f | — | — | Multi-site attachment. ∼10 chains/enzyme94 |
| Levan | Lys (n.g.)i | 75 kDa | RA | 90.5 | 0.036 | — | Multi-site attachment. Hyperbranched polymer. 50, 24, and 40 wt% enzyme in conjugate73 |
| Levan | Lys (n.g.)i | 75 kDa | RA | 76 | 0.052 | — | |
| Levan | Lys (n.g.)i | 2000 kDa, n.c. | RA | 58f | 0.095 | — | |
| (0.025)f | — | ||||||
| Polymer | Residue (number)a | MW (chain, conjugate)b | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a Number of residues modified on protein. b Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscript c (Mn) the nature of the average molecular weight was not specified. d N α-Acetyl-L-phenylalanine ethyl ester. e N α-Benzoyl-L-tyrosine-p-nitroanilide. f GlyGlyPhe-p-nitroanilide. g N α-Benzoyl-L-tyrosine ethyl ester. h Azocasein. i N-trans-cinnamoyl imidazole. j Z-GlyLeuPhe-p-nitroanilide. k GlyValPhe-p-nitroanilide. l Succ-AlaAlaProPhe-p-nitroanilide. m N α-Acetyl-L-tyrosine ethyl ester. n Bovine serum albumin. o Casein. | |||||||
| Synthetic polymers | |||||||
| PHPMA | Lys (n.g.) | 90 kDa | TCT | 0.5 | 2 | 81 | Multi-site attachment. |
| Lys (n.g.) | 53 kDa | Hyd | 1.1 | 2 | — | n.c., 3.5, and 24 wt (%) | |
| Lys (n.g.) | 13 kDa | pNP | 23.3d | 2 | 79 | Enzyme in conjugate77 | |
| (1.9)d | (68.6)d | ||||||
| MPM-06 | Lys (n.g.) | n.g. | CDI | 79e | — | — | Anionic polymer97 |
| MPEG | Lys (10) | 5 kDa | SC | — | 0.0298 | 0.180 | Effect of number of chains42 |
| Lys (14) | 5 kDa | SC | — | 0.0442 | 0.154 | ||
| (0.0433)f | (0.145)f | ||||||
| PHPMA | Lys (3) | 5.5 kDac | NHS | — | 0.055 | 0.24 | Polymeric substrate also tested50 |
| Lys (5) | 3.7 kDac | NHS | — | 0.065 | 0.27 | ||
| Lys (5) | 2.7 kDac | NHS | — | 0.05 | 0.21 | ||
| Lys(3) | 2.7 kDac | NHS | — | 0.045 | 0.18 | ||
| Asp/Glu (7) | 2.5 kDac | NHS | — | 0.3 | 0.34 | ||
| (0.39)j | (0.83)j | ||||||
| PHPMA | Lys (13) | 2.7 kDac | NHS | 72–74 | 1.807 | 0.348 | H-bonding polymer51 |
| Lys (13) | 5.1 kDac | NHS | 88 | 1.612 | 0.372 | ||
| Lys (9) | 10.9 kDac | NHS | 109–112fk | 1.364 | 0.424 | ||
| (1.052)f | (0.353)f | ||||||
| PNIPAAm-co-AADG | Lys (10) | >300 kDa | RA | 50 | 0.17 | Multi-site attachment. 14.7, 18.2, and 29.4 wt% enzyme. | |
| Lys (12) | >300 kDa | RA | 28 | 0.13 | |||
| Lys (12) | >300 kDa | RA | 31l | 0.13 | |||
| (0.12)l | Variable %AADG in polymers75 | ||||||
| PPEGMA | Lys (1) | n.c., 45.6 kDac | ABr | 86 | — | — | Grafting from. Comb-shaped polymers53 |
| Lys (4) | n.c., 52.3 kDac | ABr | 54 | — | — | ||
| Lys (8) | n.c., 90.5 kDac | ABr | 42 | — | — | ||
| MPEG | Lys (1) | 5, 32.4 kDa | NHS | 69 | — | — | |
| PPEGMA | Lys (1) | 160, 209 kDac | NHS | 57 | — | — | |
| PNaPS | Lys (4) | n.c., 78.1 kDac | ABr | 46 | — | — | |
| PPEGMA475 | Lys (4) | n.c., 52.2 kDac | ABr | 61 | — | — | |
| PDMAEMA | Lys (4) | n.c., 44.3 kDac | ABr | 38l | — | — | |
| MPEG | Lys (0.7–8) | 5 kDa | NHS | 50–85l | 0.025–0.084 | Effect of number of chains41 | |
| (0.013)l | |||||||
| MPEG | Lys (4–8) | 700 Da | NHS | — | 0.07–0.015 | 6.2–11.6 | Effect of molecular weight40 |
| Lys (1–9) | 2 kDa | NHS | — | 0.08–0.19 | 7.6–13.5 | ||
| Lys (1–8) | 5 kDa | NHS | – | 0.08–0.11 | 8.3–13.1 | ||
| (0.05)l | (8.6)l | ||||||
| Carbohydrate polymers | |||||||
| PSucr | Lys (8–11) | 400 kDa, n.c. | RA | 55–64m, 35n | — | — | Multi-site attachment. Hyperbranched polymers78 |
| PSucr | Lys (8–11) | 70 kDa, n.c. | RA | 65–75m, 26n | — | — | |
| CMC | Lys (11–13) | 12 kDa, n.c. | RA | 77–80m,80n | — | — | |
| Dextran | Lys (13,14) | 250 kDa, n.c. | RA | 50–53m, 29n | — | — | |
| CM-PβCD | Lys (4) | 13 kDa | CDI | 115m,105o | — | — | 26 wt% enzyme98 |
| Dextran | Lys (1–8) | 10 kDa | NHS | — | 0.044–0.052 | 4.3–9.4 | Single-point55 |
| (0.046)l | (9.9)l | ||||||
| Polymer | Residue (number)b | MW(chain, conjugate)c | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a Trypsin from Bovine pancreas unless otherwise mentioned. b Number of residues modified on protein. c Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscripts d (Mn) or e (Mw) the nature of the average molecular weight was not specified. f N α-Tosyl-arginine methyl ester. g Casein. h N α-Benzoyl-DL-arginine-p-nitroanilide. i N α-Benzoyl-L-arginine-ethyl ester. | |||||||
| Synthetic polymers | |||||||
| Activated PVP | Lys (n.g.) | 10, 150 kDa | NHS | 173h, 20g | — | — | Multi-site attachment103 |
| MPEG | Lys (3) | 5 kDad | TCT | 95 | — | — | Effect of number of chains45 |
| Lys (8,9) | 5 kDad | TCT | 150i | — | — | ||
| MPM-06 | Lys (n.g.) | n.g. | CDI | 70h | — | — | Anionic polymer97 |
| MPEG | Lys (12–14) | 350 Da | NPC | 154i, 366h | 0.68 | 10.0 | Effect of molecular weight43 |
| Lys (12–14) | 550 Da | NPC | 145i, 417h | 0.62 | 8.7 | ||
| Lys (12–14) | 750 Da | NPC | 145i, 390h | 0.46 | 8.6 | ||
| Lys (12–14) | 2 kDa | NPC | 154i, 355h | 0.41 | 7.2 | ||
| Lys (12–14) | 5 kDa | NPC | 154i, 364h | 0.42 | 6.5 | ||
| Lys (12–14) | 8 kDa | NPC | 127i, 293h | 0.49 | 8.2 | ||
| MPEG2 | Lys (12–14) | 2 × 5kDa | TCT | 127i, 166h | 0.37 | 2.4 | |
| (0.80)h | (2.5)h | ||||||
| PCVBPC | Lys (6,7) | 18 kDa | AzoT | 17 | — | — | Grafting from68 |
| P(MAA-co-MMA) | Lys (6,7) | 8.5 kDa | AzoT | 14g | — | — | |
| MPEG2 | Lys (7,8) | 2 × 5 kDad | NHS | 120 | 0.076 | 29.8 | Branched polymer37 |
| Lys (8,9) | 2 × 5 kDad | NHS | 125f | 0.080 | 38.5 | ||
| (0.082)f | (13.8)f | ||||||
| PNIPAAm-co-GEMA | Lys (12.9) | 37.1 kDa | CDI | 23.3 | — | — | Thermosensitive polymer104 |
| Lys (13.8) | 40.5 kDa | CDI | 27.2 | — | — | ||
| Lys (14.2) | 37.8 kDa | CDI | 14.9h | — | — | ||
| PNIPAAm | Lys (1–12) | 5 kDad | NHS | 95–124f | <KM,nath | >kcat,nath | Effect of number of chains52 |
| PDMAPS | Lys (2.4) | 11 kDa | CDI | ∼100h | <KM,nat | — | Ionic polymer95 |
| MPEG | Lys (4,5) | 1.1 kDa | CDI | 96 | 1.15 | 6.6 | Effect of coupling chemistry. Effect of molecular weight46 |
| Lys (4,5) | 2 kDa | CDI | 77 | 0.93 | 5.3 | ||
| Lys (5,6) | 5 kDa | CDI | 65 | 0.79 | 4.0 | ||
| Lys (8,9) | 1.1 kDa | TCT | 18 | 0.83 | 1.1 | ||
| Lys (7) | 2 kDa | TCT | 49 | 0.45 | 2.7 | ||
| Lys (7,8) | 5 kDa | TCT | 93 | 0.34 | 4.7 | ||
| Lys (9) | 1.1 kDa | OTs | 20 | 0.71 | 1.2 | ||
| Lys (9) | 2 kDa | OTs | 69 | 0.66 | 4.1 | ||
| Lys (9) | 5 kDa | OTs | 68h | 0.48 | 3.4 | ||
| (0.88)h | (6.1)h | ||||||
| PHPMA | Lys (n.g.) | 7 kDad | CDI | 14h | 0.14 | 0.63 | Porcine pancreas80 |
| (0.12)h | (4.6)h | ||||||
| Carbohydrate polymers | |||||||
| Dextran | Lys(n.g.) | 40 kDa | CNBr | 53f, 7g | 0.078 | — | Multi-site attachment. 11 wt% enzyme81 |
| (0.078)f | — | ||||||
| Dextran | Gln (n.g) | 72 kDa | TGase | 69i, 50g | 0.0348 | 7.5 | Multi-site attachment. 1.5, 1.8, and 0.7 chains per enzyme82 |
| CMC | Gln (n.g) | 25 kDa | TGase | 82i, 61g | 0.0150 | 9.33 | |
| PSucr | Gln (n.g) | 69 kDa | TGase | 56i, 33g | 0.0322 | 7.0 | |
| (0.0355)i | (12.3)i | ||||||
| Dextrin | Lys (n.g.) | 7.7, 39–55 kDae | CDI | 51–69 | — | 2.54–4.09 | Porcine pancreas. |
| Dextrin | Lys (n.g.) | 47, 48–123 kDae | CDI | 34–63h | — | 1.25–2.69 | Multi-site attachment79 |
| — | (2.52–2.93)h | ||||||
| Dextrin | Lys (n.g.) | 2.2, 22 kDad | CDI | 15 | 0.13 | 0.69 | Porcine pancreas. Multi-site attachment80 |
| Dextrin | Lys (n.g.) | 36, 51 kDad | CDI | 19h | 0.11 | 0.85 | |
| (0.12)h | (4.6)h | ||||||
| Polymer | Residue (number)a | MW(chain, conjugate)b | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a Number of residues modified on protein. b Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscript c (Mn) the nature of the average molecular weight was not specified. d Glycol chitin (water soluble chitin). e Micrococcus lysodeikticus. f p-Nitrophenyl tetra-N-acetyl-β-chitotetraoside. g Micrococcus Luteus. | |||||||
| P(DL-alanine) | Lys (5.8) | 1.9 kDac | NCA | 63d, 2e | — | — | Grafting from49 |
| Lys (5.7) | 1.5 kDac | 73d, 17e | — | — | |||
| Lys (5.5) | 1.2 kDac | 76d, 30e | — | — | |||
| Lys (5.4) | 1.0 kDac | 81d, 31e | — | — | |||
| Lys (3.9) | 800 Dac | 85d, 70e | — | — | |||
| Lys (3.2) | 700 Dac | 79d, 85e | — | — | |||
| MPEG | Asp (1) | 550 Da | CDI | 90 | — | — | Site specific47 |
| Asp (1) | 2 kDa | 75 | — | — | |||
| Asp (1) | 5 kDa | 38d | — | — | |||
| MPEG-deg | Lys (5,6) | 5 kDa | NHS | 0 → 60e | — | — | Degradable linker110 |
| MPEG-deg | Lys (1–6) | 5 kDa | NHS | ∼0–5 → 100e | — | — | Degradable linker111 |
| MPEG-deg | Lys (1) | 5 kDa | NHS | ∼0→∼50–95 | — | — | Several degradable linkers60 |
| MPEG-deg | Lys (5,6) | 5 kDa | NHS | ∼0→∼40–90e | — | — | |
| MPEG1200-PAsp34 | Lys (0) | ∼5.2 kDa | NC | ∼230 | — | — | Small-molecule substrate105 |
| MPEG1200-PAsp34 | Lys (3–5) | ∼5.2 kDa | GA | ∼230f | — | — | |
| PNIPAAm | Cys (1) | n.g., ∼20–40 kDa | SS and Mal | ∼100e | — | — | V131C mutant. Grafting from62 |
| MPEG | Lys (1) | 12 kDa | NHS | 2.3 → 9.6 | — | — | Certain conjugates contain cleavable linker61 |
| MPEG-deg | Lys (1) | 12 kDa | NHS | 2.8 → 97.4 | — | — | |
| MPEG-deg | Lys (1) | 20 kDa | NHS | 1.4 → 101.6 | — | — | |
| MPEG | Lys (4,5) | 12 kDa | NHS | 1.5 → 2.3 | — | — | |
| MPEG-deg | Lys (4,5) | 12 kDa | NHS | 1.5 → 95.8 | — | — | |
| MPEG-deg | Lys (3,4) | 20 kDa | NHS | 1.1 → 99.7e | — | — | |
| PVPy-b-PEO10000 | Lys (∼1) | 20 kDac | RA | 20–100e | — | — | Micellar system106 |
| MPEG | Lys (∼1) | 2.3 kDa | NHS | 80e | — | — | Ref. 59 |
| PMPC | Lys (n.g.) | 35.6 kDac | NHS + SS | ∼100g | — | — | Ionic polymer. After heating66 |
| Polymer | Residue (number)a | MW (chain, conjugate)b | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a Number of residues modified on protein. b Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscript c (Mw) the nature of the average molecular weight was not specified. d N α-Benzoyl-L-arginine-ethyl ester. e N α-Benzoyl-DL-arginine-p-nitroanilide. f Polymer conjugated to BMA block. | |||||||
| CAP | Lys (n.g.) | ∼60 kDa, n.g. | CDI | 27 | — | — | Multi-site attachment. Anionic polymer97 |
| Eudragit-S | Lys (n.g.) | ∼135 kDa, n.g. | CDI | 40 | — | — | |
| HP-55 | Lys (n.g.) | ∼45 kDa, n.g. | CDI | 17 | — | — | |
| MPM-06 | Lys (n.g.) | n.g., n.g. | CDI | 80e | — | — | |
| PSucr | Lys (n.g.) | 400 kDa, n.g. | RA | 103 | 0.94 | 0.067 | Multi-site attachment. Hyperbranched polymer83 |
| PSucr | Lys (n.g.) | 400 kDa, n.g. | RA | 97 | 1.09 | 0.059 | |
| PSucr | Lys (n.g.) | 400 kDa, n.g. | RA | 81e | 1.00 | 0.050 | |
| (1.00)e | (0.064)e | ||||||
| PMPC | Lys (7–8) | 5 kDac | NHS | 41.5 | — | — | Ionic polymer96 |
| PMPC | Lys (14–15) | 5 kDac | NHS | 34.1d | — | — | |
| PMPC | Lys (14–15) | 5 kDac | NHS | 30–35d | — | — | Ionic polymer54 |
| PMPC | Lys (12–13) | 12 kDac | NHS | ||||
| PMPC | Lys (12) | 24 kDac | NHS | ||||
| PMPC | Lys (6–7) | 37 kDac | NHS | ||||
| MPEG | Lys (12–13) | 5 kDac | n.g. | ||||
| PMPC | Lys (6–7) | 11 kDac | CDI | 41d | — | — | Amphiphilic polymer107 |
| P(MPC-ran-BMA5%) | Lys (6–7) | 10 kDac | CDI | 35d | — | — | |
| P(MPC-ran-BMA25%) | Lys (6–7) | 10 kDac | CDI | 32d | — | — | |
| P(MPC-ran-BMA50%) | Lys (6–7) | 12 kDac | CDI | 24d | — | — | |
| P(MPC-b-BM5%) | Lys (6–7) | 11 kDac | CDI | 33d | — | — | |
| P(MPC-b-BMA25%) | Lys (6–7) | 11 kDac | CDI | 33d | — | — | |
| P(MPC-b-BMA50%) | Lys (6–7) | 10 kDac | CDI | 12d | — | — | |
| P(BMA-b-MPC5%)f | Lys (6–7) | 12 kDac | CDI | 30d | — | — | |
| Polymer | Residue (number)a | MW (chain, conjugate)b | Coupling method | Activity (%) | KM/mM (native) | kcat/s−1 (native) | Comment, ref. |
|---|---|---|---|---|---|---|---|
| a Number of residues modified on protein. b Molecular weight of conjugate only given for multi-site attachment. Unless identified by superscript c (Mn) the nature of the average molecular weight was not specified. d 4-Nitrophenylacetate. | |||||||
| PNIPAAm | Cys (1) | 148 kDac | SS | 95d | — | — | Grafting from63 |
| PNIPAAm | Cys (1) | 234 kDac | Mal | 95d | — | — | Grafting from64 |
| PPEGA | Cys (1) | 7.1 kDac | VS | 92d | — | — | Partially reduced protein65 |
| PMPC | Cys (1) | 35.6 kDac | SS | ∼100d | — | — | Ionic polymer66 |
| PM13 | Lys (18) | 13 kDa, n.c. | MA | 63d | Multi-site attachment. Comb-shaped92 | ||
| PM100 | Lys (12) | 100 kDa, n.c. | MA | 93d | |||
| Polymer | Coupling | Functionality | Activity | Thermal stability | ||
|---|---|---|---|---|---|---|
| Degree of conjugation | Length of chains | Altered pH optima | ||||
| a (SP) single-point attachment; (MP) multi-point attachment; (N) neutral; (HB) hydrogen bonding; (C) charged; (×) little, no, or ambiguous effect; (+) positive effect; (–) negative effect. | ||||||
| ASNase from E. coli | ||||||
| MPEG | SP | N | −33 | −33 | ||
| MPEG2 | SP | N | −34,35 | ×74 | ||
| P(DL-alanine) | SP | N | −48 | −48 | no48 | ×48 |
| Silk fibroin | MP | N | 72 | +72 | ||
| PM13 and PM100 | MP | C | +74 | |||
| PMPEG5000-g-VP-co-MA) | MP | C | yes89 | −89 | ||
| P(VP-co-MA) | MP | C | yes90 | +90 | ||
| Sericin | MP | HB/C | yes93 | +93 | ||
| N,O-CM-Chit | MP | HB/C | yes94 | |||
| ASNase from E. carotovora | ||||||
| MPEG | SP | N | +37,38 | |||
| MPEG2 | SP | N | +37,38 | |||
| levan | MP | HB | –73 | no73 | +73 | |
| αCT | ||||||
| MPEG | SP | N | −41 | ×40 | ||
| PHPMA | SP | HB | ×50 | ×,50 +51 | no77 | +77 |
| PPEGMA | SP | N | −53 | |||
| Dextran | SP/MP | HB | −55 | ×55 | no78 | |
| PDMAEMA | SP | C | no53 | |||
| P(NIPAAm-co-AADG) | MP | HB | ×75 | |||
| PAGM | MP | HB | +76 | |||
| PNaPS | SP | C | no53 | |||
| CMC | MP | HB | no78 | |||
| PSucr | MP | HB | no78 | |||
| CM-PβCD | MP | HB | +98 | |||
| Trypsin | ||||||
| MPEG | SP | N | +43,45 | −,43,46 +46 | ||
| MPEG2 | SP | N | +37,43 | |||
| PDMAPS | no95 | |||||
| PNIPAAm | SP | N | +52 | |||
| Dextrin | MP | HB | +80 | |||
| Dextran | MP | HB | no81 | +81 | ||
| Dextran (aminated) | MP | HB/C | yes82 | +82 | ||
| CMC (aminated) | MP | HB/C | yes82 | +82 | ||
| PSucr (aminated) | MP | HB/C | yes82 | +82 | ||
| Lysozyme | ||||||
| MPEG | SP | N | −47 | |||
| P(DL-alanine) | SP | N | −49 | −49 | yes49 | |
| Papain | ||||||
| PMPC | SP | C | ×54 | ×54 | ||
| PSucr | MP | HB | no83 | +83 | ||
| CAP, Eudragit-S, HP-55, MPM-06 | MP | C | +97 | |||
2.1. Lessons learned from PEGylation
In all but one recent example, modification of L-asparaginase (ASNase) has been directed towards the amino groups on the protein, as seen in Table 1. In one of the first published examples of PEGylation of Escherichia coli (E. coli) ASNase using cyanuric chloride coupling agent, Ashihara et al.33 have modified the protein with between 18 and 77 copies of α-methoxy-poly(ethylene glycol) (MPEG) with molecular weights ranging between 750 Da and 5 kDa and have evaluated enzymatic activity using two different substrates. The authors demonstrated that both increased MPEG length and number of copies caused a decrease of enzymatic activity. A similar effect with respect to the number of chains was observed for double branched MPEG2 (2 × 5 kDa) by Matsushima et al. and Zhang et al. for E. coli ASNase.34,35 One interesting finding, which came through our analysis of PEGylated ASNase in Table 1, is that conjugates derived from Erwinia carotovora (E. carotovora) maintained higher activities than comparable conjugates prepared from E. coli ASNase. For example, Soares et al.36 have prepared a conjugate from E. coli ASNase bearing 50 copies of MPEG (5 kDa) introduced by N-hydroxysuccinimide (NHS) coupling and which has an activity of 30%, in comparison to an equivalent conjugate prepared by Monfardini et al.37 from E. carotovora and with an activity of 110%. In fact, for E. carotovora ASNase, activity was higher when modified with branched MPEG2versus linear MPEG.37,38 Of course, as ASNase obtained from E. coli and E. carotovora are serologically and biochemically different from one another,39 such differences are not surprising. In the remainder of this contribution these two proteins are treated independently. Both Monfardini et al.37 and Veronese et al.38 observed that E. carotovora ASNase coupled with either MPEG or MPEG2 possessed higher enzymatic activity than the native enzyme. This phenomenon has been observed for other enzymes (as evidenced below) and in the present case was ascribed to the production of a more active form of the enzyme given that the KM values of the modified forms were not changed upon modification.Rodríguez-Martínez et al.40 have performed a systematic evaluation of the activity of alpha-chymotrypsin (αCT) modified with 1–8 copies of NHS derivatives of MPEG with molecular weights ranging from 700 Da to 5 kDa (Table 2). The authors have found that both kcat and KM were affected by PEGylation, though these effects were independent of molecular weight. They have observed a decrease in kcat with the number of polymer chains attached concurrent with an increase in KM, which they attribute to an increase of the rigidity of the protein. This effect on KM, however, appears to be more pronounced for the shorter (≤ 2 kDa) MPEG chains versus 5 KDa chains. Castellanos et al.41 have also demonstrated that the number of copies of MPEG 5 kDa coupled to αCT correlated with decreased activity. These authors however, also showed a noticeable increase of KM with number of MPEG (5 kDa) chains attached. This trend was also observed by Chiu et al.42 who observed that the MPEG-modified αCT had well-preserved or even elevated kcat values towards low molecular weight substrates.
Gaertner et al.43 have shown that the activity of trypsin–MPEG conjugates with molecular weights ranging from 350 Da to 10 kDa increased dramatically in comparison to the native protein (Table 3). This phenomenon has been reported by others and has been attributed to a change in the local environment of the enzyme.44 Enzymatic activity, while always higher than the native (both esterase and amidase activity) decreased as a function of polymer chain length. The authors also observed a two-fold decrease of KM, while only a slight effect of MPEG molecular weight on kcat.43 The authors remarked an increase in activity of trypsin when modified with the branched MPEG2 (2 × 5 kDa) via the cyanuric chloride method. Abuchowski et al.45 have noted an increase in esterolytic activity of trypsin by increasing the number of chains of MPEG on the protein from 3 to 8–9, though a decrease of activity towards peptide and protein substrates. Monfardini et al.37 have also observed this increase of esterolytic activity upon attachment of MPEG2. Treetharnmathurot et al.46 have measured the relative activity and kinetic parameters for MPEG–trypsin conjugates prepared by three different coupling methods. All these conjugates had lower activity than the native trypsin. Within the conjugates prepared, the authors appeared to obtain dissimilar trends as a function of molecular weight between 1.1 and 5 kDa for the three different coupling strategies. That is, activity increased as a function of molecular weight when coupled using cyanuric chloride or a tosylated MPEG, but decreased when carbodiimide conjugation was used for coupling. This phenomenon is discussed in Section 3.1.
So et al.47 have observed a decrease in enzymatic activity of lysozyme–MPEG conjugates as a function of polymer molecular weight from 550 Da to 5 kDa (Table 4). The polymer was coupled in a site-specific manner to the aspartic acid residue at position 119 on the protein. This residue is distant from the protein's active catalytic site. The authors rationalize their findings by spreading of the MPEG chain over the surface of the protein resulting in interference with the protein's active site cleft.
2.2. Other polymers
2.3. General trends
Based on the findings of the previous two sections, it becomes obvious that dissimilar trends between enzymatic activity and the number/length of conjugated polymer chains are observed for the selected proteins within this contribution (Table 7). It is interesting to remark that certain proteins such as E. coli ASNase and E. carotovora ASNase display opposite trends with respect to the number and length of conjugated polymer chains, despite their related structure and function. This is an indication that the enzymatic properties of proteins are highly sensitive to polymer modification as the latter cannot only act as a diffusional barrier for the substrate, but can also modify the three-dimensional structure of the protein, thus rendering it either a more or less efficient enzyme. This also means that predicting the effect of polymer modification on the enzymatic activity of a particular enzyme is not trivial.In general, within the dataset examined, all vinyl polymers obeyed the same trend as PEGylation (albeit to a different extent) for each individual protein. This is a first indication that polymer-modification per se has an influence on protein activity. However, as highlighted by Treetharnmathurot et al.,46 coupling chemistry may also play a role on activity, notably because the authors have observed different trends with respect to molecular weight for different coupling methods (Table 7).
3. Polymer coupling strategy
With the currently available (bio)chemical toolbox for preparing protein–polymer conjugates, there are many possible ways of preparing such systems which go well beyond the random convergent coupling of a semi-telechelic polymer to a protein. The polymer coupling strategy selected for preparing the protein–polymer conjugate has a strong potential to affect the properties of the protein. This is because functional groups of diverse nature are directly added to the protein, and because these may be added in locations important for e.g. catalytic activity. Moreover, the preparative methods used to synthesize/purify the conjugates prepared by the divergent “grafting from” approach as well as the multiplicity of the bonds between a protein and a polymer can also play a key role in conjugate properties. In this section, we examine the effect of these four parameters on conjugate bioactivity. In each subsection we attempt to draw general conclusions from the available data.3.1. Ligation chemistry in the “grafting onto” approach
Within the dataset examined, there are only two studies in which several ligation chemistries are used to conjugate the terminus of a semi-telechelic polymer to a protein. Treetharnmathurot et al.46 have measured the relative activity and kinetic parameters for MPEG–trypsin conjugates prepared by three different coupling methods. All resulting conjugates had lower activities than the native trypsin. As stated previously, the authors appeared to obtain dissimilar trends as a function of molecular weight for the three different coupling strategies. That is, activity increased as a function of molecular weight when MPEG was coupled either using cyanuric chloride or a tosylated form of the polymer, but decreased when a carbodiimide was used to couple a succinoylated MPEG to the protein. Concurrently, kcat decreased as a function of molecular weight for both alkylating coupling chemistries (i.e., cyanuric chloride and tosylate derivative) but increased for the acylating coupling chemistry (carbodiimide). The KM of all conjugates decreased as a function of molecular weight. From this study it appears as though, for trypsin, ligation chemistry which maintains the charge of the ε-amino groups of the lysine residues on the protein (albeit with a potential modification of their pKa), has a positive effect on enzymatic activity. Lu et al.50 have modified αCT with semi-telechelic PHPMA either at lysine residues or at aspartic/glutamic acid residues (Table 2). Conjugates prepared by modification of the amino groups on αCT showed higher activities than the native enzyme. However, the authors have observed a decrease of enzymatic activity for equivalent conjugates for which the PHPMA chains are conjugated to carboxylic acid groups on the protein.50 It is known that a carboxylic acid group of an aspartic acid is involved in the active site of αCT.56 With the limited number of systematic studies available to discuss the effect of conjugation chemistry, it is impossible to draw more general conclusions at this time.3.2. Random versus site-specific coupling
Site-specificity is an interesting and modern approach for preparing conjugates with potentially less loss of enzymatic activity.30 In this approach, detailed knowledge of the protein's three-dimensional structure is required in order to select locations for polymer-modification far from its catalytic site. Alternatively, one can select a location near or within an antigenic region of the protein in order for the conjugate not to be recognized by the immune system. In this section, we highlight the benefits or disadvantages of site-specific protein modification in comparison to random modification. Of course, as such modifications are specific to a given protein, the latter are treated individually.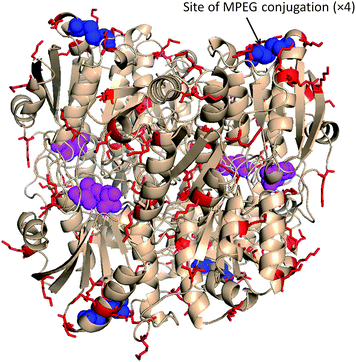 | ||
| Fig. 1 Tetrameric L-asparaginase from E. coli (DPB entry 4ECA). Thr-12 in active region of each monomer acetylated with L-aspartic acid (magenta). Site-specific introduction of the polymer chains at Cys77–Cys105 (blue) yielded a conjugate with full retention of activity, but inefficient elimination of antigenicity.58 Protein image produced with PyMol v.1.1. | ||
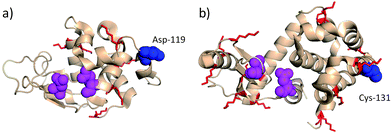 | ||
| Fig. 2 (a) Hen egg lysozyme (PDB entry 2HU1) with residues implicated in catalysis (Glu-35 and Asp-52) in magenta and Asp-119 in blue. (b) T4 lysozyme V131C (PDB entry 2HUK) with residues implicated in catalysis (Glu-11 and Asp-20) in magenta and Cys-131 in blue. For both proteins, lysine residues are in red. Conjugation at Cys-131 lead to full retention of enzymatic activity (antigenicity not tested). Modification at Asp-119 lead to partial loss of activity and did not eliminate antigenicity.47,62 Protein images produced with PyMol v.1.1. | ||
Overall the examples seen in this section demonstrate that the location chosen for protein modification is crucial for obtaining a conjugate with desired properties. Indeed, when the location is properly chosen, the characteristics of the polymer, within the dataset examined, appear to play a secondary role on bioactivity.
3.3. “Grafting from”
The so-called “grafting from” approach for preparing protein–polymer conjugates by direct polymerization from a protein is associated with many advantages, including: the possibility of accurately characterizing the sites of polymer modification, the ease of purification of the conjugate, and the greater uniformity of the produced conjugates.18,23,27 However, in order for the “grafting from” approach to be viable, the protein must survive modification with the initiating moiety, the polymerization reaction (which can involve transition metals or ionizing radiation), and the purification procedure without compromising significantly activity. In this section, we discuss aspects of the synthesis, the purification, and the opportunities offered by this approach, in particular in as much as how they affect enzymatic activity.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dimethyl sulfoxide:aqueous buffer. Depending on the desired characteristics of the conjugate, either 100 mM K2HPO4 (pH 6.8, 0 °C) or 50 mM NaHCO3 (pH 8.6, 4 °C) was used. Yoshimura et al.49 have encountered a similar problem for the preparation of conjugates of lysozyme with the same polymer. In this case, the authors performed their polymerizations in 20 vol% dioxane in 50 mM phosphate buffer (pH 7.1, 0 °C). In both previous examples, the proteins maintained reasonable enzymatic activity following exposure to organic solvents. In particular, conjugates prepared with lysozyme maintained activity towards glycol chitin to a similar (or even better) extent to conjugates prepared with MPEG (Table 4) by other authors. This is an indication that, to a certain extent, organic solvents are acceptable for improving the homogeneity of the polymerization medium without drastically affecting the enzymatic activity of the conjugate.
:1 dimethyl sulfoxide:aqueous buffer. Depending on the desired characteristics of the conjugate, either 100 mM K2HPO4 (pH 6.8, 0 °C) or 50 mM NaHCO3 (pH 8.6, 4 °C) was used. Yoshimura et al.49 have encountered a similar problem for the preparation of conjugates of lysozyme with the same polymer. In this case, the authors performed their polymerizations in 20 vol% dioxane in 50 mM phosphate buffer (pH 7.1, 0 °C). In both previous examples, the proteins maintained reasonable enzymatic activity following exposure to organic solvents. In particular, conjugates prepared with lysozyme maintained activity towards glycol chitin to a similar (or even better) extent to conjugates prepared with MPEG (Table 4) by other authors. This is an indication that, to a certain extent, organic solvents are acceptable for improving the homogeneity of the polymerization medium without drastically affecting the enzymatic activity of the conjugate.
Overall, the examples discussed in this section demonstrate that it is possible to maintain high levels of protein bioactivity following synthesis and purification of conjugates prepared by the “grafting from” approach.
3.4. Multi-point covalent bonding (reactive polymer side-chains)
In the previous sections, the modification of proteins with semi-telechelic polymers has been discussed. However, the multi-point attachment of enzymes to polymers (i.e., the polymer bears multiple protein-reactive groups along its backbone) has been widely studied and is considered to be the most general approach to stabilize these proteins against different denaturing conditions,70 in particular for avoiding their inactivation caused by protein unfolding. This holds particularly true for enzymes composed of multiple subunits, such as ASNase, and whose enzymatic activity is lost due to dissociation into inactive subunits through dilution.71 Consequently, and as highlighted in this section, multi-point attachment has the potential to increase the thermal stability of the conjugate because of its preventative effect on protein denaturation as well as to affect the pH-activity profiles of the conjugate because of the change in the microenvironment surrounding the enzyme.In the multi-point attachment approach, because both the polymer and the protein possess multiple functional groups susceptible to react together, the resulting conjugates are, in general, less well-defined than their analogs prepared by single-point attachment. One ambiguity which must be clarified is that, in contrast to conjugates prepared by single-point attachment, the degree of modification of the reactive residues on the protein is not necessarily proportional to the number of conjugated polymer chains. As a consequence, for these conjugates the molecular weight of the final conjugate is a second necessary parameter for describing the conjugate. Unfortunately, for many of the conjugates listed in Tables 1–6, this important parameter is not defined and thus analysis of the influence of polymer attachment is somewhat hampered. In this section we discuss the effect of multiple covalent coupling of proteins to polymers and do not discuss any specific non-covalent interactions between the two. We have, however, included in this section polymers bearing hydroxyl groups, which have the potential to participate in hydrogen bonding with the protein, because we found it was not possible to distinguish the effect of this type of non-covalent interaction from that of multiple covalent coupling.
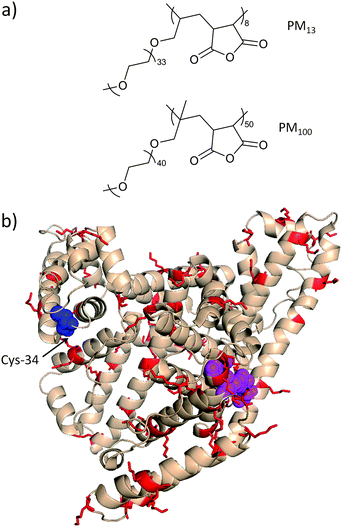 | ||
| Fig. 3 (a) Comb-type polymers developed by Inada and co-workers for multi-point attachment to ASNase and BSA.74,92 (b) Human serum albumin (PDB entry 1AO6) with residues implicated in esterase activity in (Asn-391, Arg-410, and Tyr-411) magenta, and Cys-34 in blue. Several semi-telechelic polymers have been site-selectively conjugated to Cys-34 of bovine serum albumin (pdb structure not available) with excellent retention of activity. Multi-point attachment of comb-type polymers PM13 and PM100 to lysine residues (red) lead to a moderate decrease in activity. Protein image produced with PyMol v.1.1. | ||
Overall, it can be seen in this section that careful choice of synthetic conditions is required for obtaining a reasonably well-defined conjugate. Multi-point attachment of polymers to proteins is an effective means of improving their thermal stability. In the preceding examples, little mention is made of the individual contribution of covalent bonding versus hydrogen bonding on enzyme properties. However, as the polymers in this section are neutral, little or no effect on pH-dependant activity profiles was observed (Table 7). Also, it becomes apparent that the degree of covalent coupling alone cannot account for all observed trends, indicating that polymer properties (such as polarity, architecture, etc.) and the susceptibility of the latter to interact with the protein/substrate play key roles in the enzymatic properties of the conjugate. These will be discussed in turn in the following two sections.
4. Polymer architecture
In this section we survey the influence of polymer backbone architecture on the catalytic properties of protein–polymer conjugates. It is generally known that branched or folded polymers adopt more compact structures than linear polymers in a random-coil configuration. Therefore these polymers may possess a smaller “footprint” on the protein's surface and potentially confer different properties to a conjugate than a flexible linear polymer, which can spread on the protein's surface. Consequently, the way they perform as a diffusion barrier to substrate is likely to be different from that of linear polymers. In this section, we will examine the influence of polymer backbone architecture or propensity to adopt a distinct conformation on conjugate enzymatic properties. We will sequentially discuss polysaccharides which may be linear or hyperbranched, polypeptides with folded structures, and finally comb-type synthetic polymers. Of worthy mention, while MPEG2 constitutes an elementary branched polymer (from the perspective of the enzyme), the lack of studies which systematically evaluate the length of the polymer segments, the number of conjugated MPEG2 chains, or the degree of branching (i.e., higher than 2 through an alternative coupling strategy to TCT) unfortunately limits what can be said about the effect of this type of branched polymer on conjugate bioactivity. Comparisons of MPEG2 to MPEG have already been made in Section 2.1. and will not be treated further.4.1. Polysaccharides
Polysaccharides have the potential to display different degrees of branching, depending on their source. For instance, CMC is a purely linear polymer, while dextran and dextrin possess a certain non-negligible degree of branching, and bacterial levan and polymeric sucrose are highly branched and adopt an almost spherical structure in solution.84,85 For a polymer of a given molecular weight, hyperbranched macromolecules adopt more compact and rigid structures in solution than flexible linear polymers. In addition, hyperbranched polymers do not interpenetrate.86 Therefore, for an enzyme modified with several polymer chains, these are less likely to interpenetrate to form a uniform diffusion barrier to small molecule substrates, in comparison to a polymer adopting a loose random coil configuration. Clearly, architecture-dependent properties are expected between these classes of polymers. Unfortunately, very little or no information is given in the studies reviewed herein on the degree of branching of these polymers, owing to the difficulties in characterizing this aspect of natural polymers. As such, useful comparisons of the conjugates are difficult to make.Sundaram and Venkatesh78 have evaluated the thermal stability of native and variously modified αCT with polysaccharides including polymeric sucrose (70 and 400 kDa), CMC (12 kDa), and dextran (73 and 250 kDa). The difference in thermal stability is rather unusual in that it decreased from the highly branched polymeric sucrose, to the linear CMC, to the branched dextran. The authors demonstrated that polymer size alone is not the influential factor in improving the stability of a protein, its structure obviously plays an important role, as confirmed by their finding that polymeric sucrose (70 kDa) and dextran (73 kDa) stabilized αCT to different extents though they have about the same molecular mass. Unfortunately, as the molecular weights of the conjugates have not been evaluated, it is not possible to draw more detailed conclusions from this study. The authors rationalized their findings in that the flat ribbon-like chains of CMC would be the most rigid of the three polymers used, based on the suggested interpretation that bulky groups in carbohydrates equatorial to the glycosidic linkage make them so. Additionally, the rigidity of the highly branched polymeric sucrose may also contribute to the enhanced thermal stability of the conjugate. From the measured activity of the conjugates presented in Table 2, the conjugates prepared with a comparable degree of modification of αCT (11–13 and 13–14 lysine residues for CMC and dextran, respectively) only show a small decrease in activity when going from 12 kDa CMC to 250 kDa dextran. In comparison, attachment of a single 5 kDa MPEG chain to αCT by Lele et al.53 led to a comparable degree of inactivation. Also of note is the fact that the conjugate prepared with polymeric sucrose (8–11 reacted lysine residues, 70 kDa per chain) had the same activity as the conjugated prepared with CMC above. Furthermore, the conjugate prepared with 400 kDa polymeric sucrose had higher activity than the conjugate prepared with 200 kDa dextran, indicating that in this case, dextran was more effective than polymeric sucrose in blocking the enzyme's active site. Additional characterization of these conjugates would be necessary to unambiguously establish the reason for this.
In a comparable study, Villalonga et al.82 have modified trypsin with dextran (72 kDa), CMC (25 kDa), and polymeric sucrose (69 kDa) by means of the enzyme transglutaminase (TGase). For this coupling reaction, the polysaccharides were oxidized and amino groups introduced with diaminobutane/NaBH4. These amino groups act as acyl acceptors for the TGase-mediated reaction with γ-carboxyamide groups on the protein. They have noticed that lower thermal stability was conferred to the enzyme after glycosidation with CMC, in comparison with the dextran and polymeric sucrose complexes. No clear differences were observed between the low to moderately branched dextran and highly branched polymeric sucrose. However, the conjugate prepared with polymeric sucrose had a lower proteolytic activity in comparison to the dextran conjugate, despite a lower level of protein modification. In contrast, the esterolytic activity appeared to be less affected. This is in contradiction with the results from the preceding paragraph and points to the need for better characterization of conjugates in order to draw meaningful conclusions for the effect of polymer architecture. The lower thermal stability of the conjugate prepared with CMC was rationalized to result from the conformation of CMC, which adopts a flat ribbon-like structure in solution, thereby causing less interactions (and possibly lower extents of multi-point attachment) in comparison to the more flexible branched dextran or the more-or-less spherical polymeric sucrose.
4.2. Polypeptides
Polysaccharides are not the only polymers to display specific backbone configurations. Indeed, proteins are (in general) linear polypeptides which can adopt compact folded structures with little potential for interpenetration. There is only one example, within the dataset examined, of a conjugate prepared with a folded polypeptide. Poznansky et al.87 have modified E. coli ASNase with rat albumin via a multi-point attachment approach using gluteraldehyde, and have managed to maintain activity to 60% of the native protein, despite the presence of an average of 12 large (∼65 kDa) albumin molecules per enzyme (Table 1). However, performing the modification reaction in absence of the substrate L-asparagine resulted in the loss of at least 90% of the enzyme activity. The authors indicated little change in Vmax, a 2- to 3-fold increase of KM, increased thermal stability, and resistance to proteolytic digestion. It is possible that, in contrast to common synthetic polymers, which adopt random coil conformations in solution, the grafted albumin molecules maintain their compact folded structure which would prevent the approach of large molecules such as antibodies but allow the approach of smaller molecular substrates.4.3. Comb-type polymer backbones
Comb-type polymers possess interesting characteristics because they are more well-defined than the hyperbranched polysaccharides discussed above. In addition, comb density, the length of the combs, and the bulkiness of the combs can be adjusted to confer different properties to the polymer and consequently also the conjugate. While indeed partial stretching of the main-chain of comb-type polymers is generally observed, the chains generally adopt conformations that render the overall system more compact than that adopted by linear polymers.88 In this section, we survey the effect of comb density and comb length on conjugate properties given that, in all examples available, the chemical nature of the comb (MPEG) is the same.In two separate studies, Ma and co-workers89,90 have evaluated the effect of modification of ASNase with either linear P(VP-co-MA) or comb-shaped P(MPEG5000-g-VP-co-MA) (MA: maleic anhydride), that is, with or without polymeric side-chains. The authors observed that the degree of protein modification required for elimination of the antigenicity of ASNase decreased as the proportion of MPEG side-chains on the comb-type polymer increased. Complete elimination of antigenicity of ASNase was achieved by modification of 63 amino groups with linear P(VP-co-MA) in comparison to 30 amino groups for the comb-shaped P(MPEG5000-g-VP-co-MA). For comparison, complete elimination of antigenicity of ASNase modified with either linear or double branched MPEG2 (2 × 5 kDa) is achieved only at higher degrees of enzyme modification and only low enzymatic activity is retained.33,91 Interestingly, the comb-shaped polymer had a lower optimal activity temperature in comparison to the native enzyme, which may indicate that multi-point attachment between the protein and polymer is disfavored due to the bulky MPEG combs. Additionally, enzymatic activity remained higher when the polymer possessed a higher proportion of MPEG side-chains. Unfortunately, as the molecular weight of the conjugates have not been characterized in this study, it is not possible to further explain these results as it is unclear whether the number of polymer chains per ASNase molecule is altered as a function of increasing comb density.
Inada and co-workers74,92 have modified both E. coli ASNase and BSA with both PM13 and PM100, which are comb-type polymers with 1.5–2 kDa MPEG side-chains and with overall molecular weights of 13 and 100 kDa, respectively (Fig. 3a). For ASNase, the authors observed that polymer molecular weight was an influential factor on antigenicity. That is, at a degree of modification at which no antigenicity of the conjugate was observed 46 and 30 lysine residues were modified by PM13 and PM100, respectively. This trend was also consistent for BSA for which antigenicity was lost following modification of 18 or 12 lysine residues, respectively (Fig. 3b). For both proteins, enzymatic activity was higher for PM100 than for PM13 or MPEG2 (Table 1). Interestingly, and in contrast to the results of Ma and co-workers,89,90 the conjugates had greater stability towards heat and denaturing conditions, and this as a function of polymer molecular weight. This result may perhaps stem from the shorter combs (i.e., 1.5–2 kDa) in PM13 and PM100 compared to the 5 kDa combs on the polymers of Ma and co-workers, which may have led to greater multi-point attachment. One could equally argue that the difference in length of the two combs is much less pronounced than the difference in the molecular weight of the two polymers, which could also be the dominant factor here.89,90
Other comb-type conjugates with much smaller side-chains have been prepared by Lele et al.53 by direct polymerization of PEGMA from a αCT macro-initiator. Increase of the molecular weight of the PPEGMA chain from ∼19 kDa to 160 kDa had only a small effect on activity, which indicates that individual comb-type polymer chains impose only a small diffusion barrier to small molecules. This may be an indication that the polymer is adopting a specific conformation that does not allow it to spread over the surface of the protein and interfere with its active site. In another example, Grover et al.65 have modified bovine serum albumin with a single 7.1 kDa chain of PPEGA and have observed only a slight decrease in activity. This result should, however, be considered in the light of our previous comments on the enzymatic activity of BSA. Unfortunately, in both preceding examples, thermal stability and antigenicity have not been evaluated for comparison with the other conjugates from this section.
4.4. General trends
Based on the available literature it is not clear in what manner polymer backbone architecture can be exploited to enhance thermal stability and suppress antigenicity/proteolysis, without compromising enzymatic activity. It appears as though polymers with rigid backbones resulting either from branching, folding, or the presence of bulky side-chains influence to a lesser extent enzymatic activity of the conjugate than flexible linear polymers. This may be due to the lower ability of these polymers to spread over the surface of the enzyme and interfere with active regions. Also, the rigidity of the polymer may confer enhanced resistance to denaturation by heat as the polymer is likely to convey this property to the enzyme, thus stabilizing its structure and reducing its potential for denaturation, especially in the case of concurrent multi-point attachment.5. Polymer side-chain functionality
In contrast to PEG, which is generally considered to be chemically inert under pharmaceutically relevant conditions, most of the natural and synthetic polymers in Tables 1–6 possess side-chains that are not inert and can thus interact either amongst themselves, with the solvent, with the protein, or with the enzymatic substrate. That is, non-covalent interactions such as hydrogen bonding (e.g., with polysaccharides, PHPMA, poly(N-vinylpyrrolidone) (PVP), etc.), electrostatic interactions (e.g., between positively charged lysine residues and ring-opened maleic anhydride repeat units), host–guest interactions (e.g., between the polymer and the protein, substrate, etc.), or other uncharacterized interactions also contribute to the overall properties of the enzyme. In this section, we successively discuss electrostatic interactions, host–guest interactions, and other less well-characterized interactions between a polymer and the enzyme or its enzymatic substrate and highlight the influence of these interactions on the bioactivity of the conjugate. We have found that it was not possible to distinguish properties related to hydrogen bonding from those stemming from covalent coupling and therefore have treated these together in Section 3.4. The specific effects discussed herein can be distinguished from polymer conjugation per se. Also, in certain cases, the authors have performed specific control experiments to identify the specificity of the phenomenon. Of worthy mention, in this section it is not unambiguous to distinguish whether the effect on bioactivity results from interaction between the polymer and the enzyme, between the polymer and the substrate, or to another uncharacterized phenomenon. In addition, the observed effects in subsection 5.3. tend to be relatively specific to a given polymer/enzyme/substrate system. Nevertheless, as ultimately the number of different polymers and enzymes typically used to prepare protein–polymer conjugates remains small, specific interactions such as those to be discussed in this section remain important and merit mention.5.1. Electrostatic interactions
In Section 3.4 we have shown that neutral polymers conjugated to enzymes via multi-point attachment enhanced the thermal stability of the conjugate without altering its pH-activity profiles (Table 7). However, we have observed that in many instances where the polymer possessed a charge, pH-activity profiles of the conjugate were indeed altered (Table 7), indicating that in addition to the effects concurrent with polymer modification previously discussed, ionic interactions, whether between the polymer/enzyme or polymer/substrate, can also play a role in the characteristics of the conjugate. For example, Zhang et al.93 have modified ASNase with sericin, a water-soluble polypeptide with molecular masses ranging from 20 to 60 kDa and composed of 15 types of amino acids, among which polar amino acids with hydroxyl, carboxyl, and amino groups such as serine, aspartic acid, and lysine account for 72% of all residues. The optimal operating pH of the resulting conjugate shifts considerably to 5 in comparison with pH 6–8 of the native form of the enzyme. In addition, the thermostability and resistance to trypsin digestion of the modified enzyme were greatly enhanced as compared with ASNase alone owing to the multi-point covalent coupling approach used to prepare the conjugate. Ma and co-workers89,90 have modified ASNase with copolymers of PVP and maleic anhydride, both with and without pendant MPEG chains (i.e., comb-shaped architecture, discussed in Section 4.3). The pH optima of enzymatic activity of the modified ASNase was slightly more alkaline than the native enzyme (shifted by 0.4 and 0.8 units higher for copolymer with and without MPEG, respectively) which the authors attribute to the anionic microenvironment surrounding the enzyme. Qian et al.94 have modified E. coli ASNase with N,O-carboxymethyl chitosan and have also noted that the modified enzyme showed slightly more alkaline pH optima than did the native enzyme. This may result from the charged nature of the polymer.Lele et al.53 have prepared conjugates of αCT by polymerization of sodium 4-styrenesulfonate (NaPS) and N,N-dimethylaminoethyl methacrylate (DMAEMA) from the protein macro-initiator. Interestingly, and in contrast to the preceding results for ASNase, comparable enzymatic activities are observed despite the significantly different structure and properties of these two polymers, indicative that for these particular systems the net charge of the conjugate is not a determining factor for enzymatic activity (at the tested pH). Based on this limited dataset, it is not possible to comment on whether this is a general phenomenon for αCT or whether it is simply valid for the above-mentioned conjugates.
Yan et al.95 have coupled the thermosensitive zwitterionic polymer poly(3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) (PDMAPS) to trypsin. The PDMAPS and the conjugate have their upper critical solution temperature at 14 and 16 °C, respectively. Investigation of the fluorescence of the tryptophan residues on the protein suggested that the conjugation did not lead to any significant change in the microenvironment of the tryptophan residues of the trypsin. In other words, the conjugated trypsin remained in its native tertiary conformation. Circular dichroism spectroscopy indicated that α-helical and β-sheet content of the conjugate were both slightly reduced. The optimal pH for activity was unchanged after conjugation, which the authors attribute to the fact that at pH 7 PDMAPS is neutral due to charge pairing. The conjugate had a smaller KM as compared to that of the native trypsin, possibly due, as suggested by the authors, to enhanced hydrophobicity for the conjugated trypsin. Interestingly, the optimal temperature for proteolysis of casein of the conjugates increased from 20 °C (native) to 60 °C (conjugate), concurrent with a large increase of activity (well above the 100% activity of the native enzyme), though the reason for this is unclear.
In another example of conjugates prepared with zwitterionic polymers, Ishihara and co-workers54,96 have modified papain with PMPC and have observed that the helix content of conjugated papain was slightly below that of native papain, indicating that conjugation slightly altered enzyme structure as a function of both the number of chains and polymer molecular weight. This change in enzyme conformation did not affect enzymatic activity as all conjugates, including a MPEG conjugate, had similar esterolytic activities (ca., 30–35%, Table 6). Unfortunately, the pH-dependent activity profile for these conjugates was not measured. Fujimura et al.97 have covalently modified papain with different enteric coating polymers by means of carbodiimide coupling to the pendant carboxylic acid side-chains on the polymer. Heat stability of the resulting conjugates increased compared with that of native papain, and the conjugate had higher stability against water-miscible organic solvents and protein denaturing agents likely due to multi-point attachment. However, as the polymer itself is pH-sensitive, assessing the influence of the electrostatic interactions between the protein and the polymer on the optimum pH for activity is meaningless.
5.2. Host–guest interactions
Supramolecular interactions represent an interesting class of non-covalent interactions useful for stabilizing proteins. For instance, Fernandez et al.98 have modified αCT with O-carboxymethyl-poly-β-cyclodextrin (Fig. 4) by coupling the pendant carboxylic acid groups to amino groups on the protein. This polymer has the potential to form stable inclusion complexes between cyclodextrin (βCD) units and hydrophobic compounds such as the substrate or certain amino acid residues located at the surface of the enzyme. The authors have found that the attached polymer served to maintain the active conformation of the enzyme after thermal treatment principally through covalent multi-point attachment of the polymer chains (via carbodiimide coupling), though multi-point salt bridges resulting from the polyanionic nature of the modifying polysaccharide, as well as through supramolecular interactions involving hydrophobic amino acid residues and the βCDs were speculated to contribute as well. This last phenomenon was verified by addition of 1-adamantanol, which forms a highly stable 1:1 complex with βCDs, resulting in a reduction of the protective effect of the polymer on the thermal resistance of the modified conjugate at 50 °C. Moreover, the half-life of the modified enzyme was only 1.5-fold increased in the presence of the guest 1-adamantanol with respect to the native counterpart, in contrast to an increase of about ∼12-fold without addition of the guest. The stabilization of enzymes through host–guest interactions by modification with βCDs has also been reported by others.99–101 The thermal stabilization of trypsin using a βCD-modified dextran as additive (i.e., not covalently coupled to trypsin) has also been reported.102
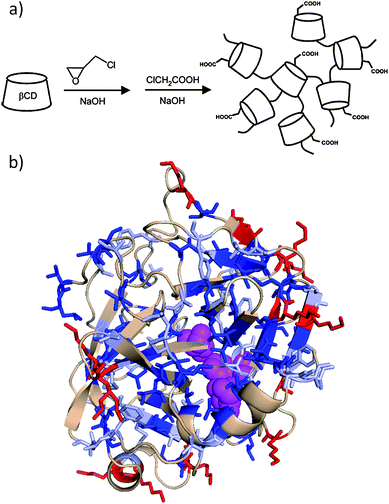 | ||
| Fig. 4 (a) A schematic representation of O-carboxymethyl-poly-β-cyclodextrin prepared by polymerization of β-cyclodextrin (βCD) with epichlorohydrin.98 (b) The monomeric form of bovine alpha-chymotrypsin (PDB entry 1YPH) with hydrophobic amino acids in blue (Val, Ile, and Leu dark; Ala, Phe, Cys, Met light), lysine residues in red, and the active catalytic site (His-57, Asp-102, and Ser-195) in magenta. The polymer in (a) was conjugated to the lysine residues on α-chymotrypsin. Host–guest interactions between the β-cyclodextrin units and hydrophobic amino acid residues on the surface of the enzyme resulted in enhanced stability of the conjugate towards heat. Part (a) is adapted from ref. 98. Protein image produced with PyMol v.1.1. | ||
5.3. Other interactions or effects
In addition to the specific examples above, the functional groups along the backbone of the polymer may influence the polarity, diffusivity, etc. of the microenvironment surrounding the enzyme in a less precise manner, thus affecting how the substrate interacts with the latter. In this subsection we summarize examples in which the nature of the polymer has a distinct effect on enzymatic activity of the conjugate, whether this underlying phenomenon has been elucidated or not.von Specht and Brendel103 have covalently modified trypsin with PVP by multi-point attachment by prior opening of a certain percentage of the lactam side-chains. The resulting conjugate showed an increase in specific activity compared to the native enzyme towards low molecular weight substrates. However, activity towards large substrates such as azocasein was much reduced. This difference of activity indicates that the main changes which are caused by binding short chains of PVP are of steric origin. The authors attributed the increased activity of trypsin towards small substrates to an increased local concentration of the latter stemming from the known binding ability of PVP towards small molecule dyes and drugs. Ding et al.52 have prepared conjugates of trypsin and semi-telechelic PNIPAAm. The authors observed that the esterase activity of the conjugates increased in relation to that of the native protein. Furthermore an increase of activity with the extent of polymer conjugation was observed. These effects were also observed in the amidase activity of the conjugate. The authors speculated that this could be related to the change in microenvironment of the enzyme's active site, leading to enhanced partitioning of substrate in the polymer phase. Lee and Park104 have prepared conjugates of trypsin with a semi-telechelic copolymer composed of NIPAAm and a glucose monomer and bearing a terminal carboxylic acid group, which was used for coupling to protein. By considering the weight proportion of enzyme within the conjugate, the amidase activity of the enzymes appeared to be enhanced compared to the native enzyme. Interestingly, the conjugated enzymes demonstrated very peculiar enzyme activity-temperature profiles, with two apparent optimal temperatures, indicating that both temperature-controlled collapse and flocculation of the copolymers around the enzyme surface modulated the mass transfer rates of substrate to the active site of the enzyme.
Interestingly, lysozyme modified with 35.6 kDa PMPC in a random fashion exhibited ∼100% activity towards the macromolecular substrate Micrococcus Luteus, which contrasts with the known deactivation of this enzyme upon random conjugation of a polymer to lysine residues (Table 4).66 Whether this is a specific effect of the zwitterionic polymer or due to the choice of a different substrate (M. Lysodeikticus is a more generally used substrate in Table 4) cannot be established herein. Yuan et al.105 have exploited the self-assembly characteristics of a lysozyme/MPEG–poly(aspartic acid)) (MPEG5000-PASP34) mixture to form polyion complex (PIC) micelles, which were subsequently cross-linked by addition of gluteraldehyde. This step promoted reaction between the amino groups of lysozyme and the ω-amino end-group of the polymer within the core of the micelle. The authors have demonstrated the effect of substrate sequestering on the enzymatic activity towards a synthetic small-molecule substrate able to penetrate into the core of the micelle. In comparison to the native enzyme, the conjugates displayed significantly enhanced activity, independently of the amount of gluteraldehyde added for cross-linking indicating that, within the experiments performed, core-cross-linking did not inhibit the enzymatic activity of the entrapped lysozyme. Danial et al.106 have prepared complex coacervate core micelles with a lysozyme-modified corona by coupling the latter to the end of a poly(2-vinyl pyridine)-b-PEG block copolymer. The enzymatic activity of the lysozyme-micelle conjugates towards M. lysodeikticus was comparable to that of free lysozyme, though decreased when the number of enzyme molecules per micelle increased. This was attributed to sequestration of a certain number of enzymes within the core of the micelle, where interaction with the substrate was hindered.
Miyamoto et al.107 have increased the hydrophobicity of PMPC by copolymerization with n-butyl methacrylate (BMA) in differing proportions and have conjugated the resulting polymers to papain (Fig. 5). The authors observed a loss of α-helical content of the papain only for the most hydrophobic block copolymer prepared from these two monomers. Concurrently, enzymatic activity decreased with increasing amounts of BMA in the copolymer, which might be due to enhanced restriction of substrate diffusion to the enzyme. These polymers nevertheless stabilized the protein due to prevention of auto-digestion.
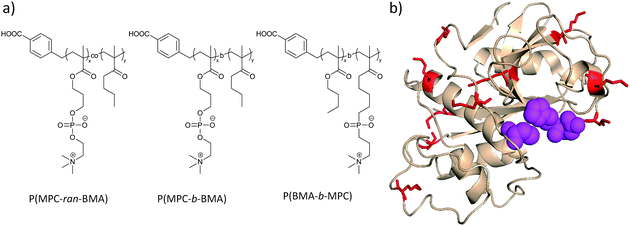 | ||
| Fig. 5 (a) Copolymers of the zwitterionic monomer MPC with the hydrophobic monomer BMA were conjugated to lysine residues (red) on papain (b, PDB entry 1PPP). The active site of the enzyme (Cys-25, His-159, and Asp-158) is shown in magenta. The activity of the enzyme decrease with increased BMA content.107 Protein image produced with PyMol v.1.1. | ||
5.4. General trends
Within the examples examined, it appears as though polymers bearing charged functional side-chains affect the pH-dependent activity profiles of enzymes such as ASNase and trypsin. This was not the case, however, for αCT whose pH-dependent activity was not affected by conjugation of charged polymers. Interestingly, the zwitterionic polymer PDMAPS did not alter the pH optima of activity of trypsin. Host–guest interactions between polymeric βCDs and hydrophobic residues on αCT or trypsin appear to effectively improve thermal stability, whether the polymer is covalently coupled to the enzyme or not. This phenomenon is readily distinguished from all other interactions by addition of 1-adamantanol. The last subsection has highlighted that the functional side-chains of the polymer can contribute to phenomena which have a very significant effect on bioactivity. This effect can in fact lead to a pronounced improvement of catalytic activity in comparison to the native enzyme. Overall, this section demonstrates that polymer side-chains functionality must be considered either in conjugate design (for the limited trends above) or when interpreting conjugate properties.6. Polymer biodegradability
Biodegradability is an important feature of protein–polymer conjugates designed for use in humans. Increasing the molecular weight of therapeutic proteins through polymer conjugation is an easy way of avoiding clearance by the kidneys, though accumulation of large, non-biodegradable components in the liver may lead to undesirable side-effects. Understanding the mechanism through which the protein–polymer conjugate will decompose over time is important for understanding how its activity will evolve over this period, and for predicting its ultimate fate in the body. Biodegradability can be achieved in a number of ways.108 In the simplest case, the polymer can be biodegradable along its main-chain, leading to a progressive reduction in molecular weight and production of small molecules. Polypeptides and polysaccharides are examples of such polymers that may degrade under appropriate hydrolytic and/or enzymatic conditions. Alternatively, a non-biodegradable polymer may be conjugated to the enzyme via a labile linker, which, upon biodegradation, releases the intact polymer chains. In this case, care must be taken that the individual polymer chains have molecular weights low enough to be eliminated by the kidneys. Conjugates containing MPEG coupled to the protein via a cleavable linker are examples of such systems. To a certain extent, all polymers linked to the protein via a potentially labile bond, such as an ester or an amide bond, also fall into this category. Lastly, the polymer may hydrolyze along its side-chain, thus progressively reducing the molecular weight. Polymers with large side-chains such as ca. PM13 or PM100 fall into this category as release of high molecular MPEG upon hydrolysis of the ester bond which binds it to the main-chain would dramatically reduce the weight of the parent polymer. In this section, we discuss the effect of these three forms of biodegradation on enzymatic activity.6.1. Main-chain biodegradable polymers
Polypeptides are susceptible to proteolysis and therefore fall into the category of main-chain biodegradable polymers. Within the literature examined herein, no specific mention is made of attempting to selectively remove polypeptides covalently coupled to enzymes. However, in certain cases, the stability of the conjugates in the presence of proteolytic enzymes is reported and the enzymatic activity reported as a function of time. This experiment indirectly examines the ability to selectively degrade the conjugated polypeptide chains versus the enzyme within its core. For instance, Uren and Ragin48 have prepared conjugates of poly(DL-alanine) with ASNases and have evaluated different proteolytic conditions for assaying conjugate stability. The authors observed that, over a one hour period, the conjugate was totally resistant to trypsin, while 50% of enzyme activity was lost for the native protein. Therefore, under these conditions, any possible proteolysis of poly(DL-alanine) had little or no effect on the activity of the conjugate. In contrast, upon incubation of the conjugate with αCT, enzymatic activity was reduced, though to a significantly lesser extent that for native ASNase. Given that, a priori, poly(DL-alanine) does not contain aromatic residues which are the typical substrate for αCT, it is possible that the primary effect observed is the retarded degradation of ASNase due to the imposed steric barrier of the polymer. These results indicate that it is not possible to selectively degrade the poly(DL-alanine) with αCT without compromising ASNase activity. In another example, Poznansky et al.87 have evaluated the proteolytic stability of ASNase modified with albumin, a polypeptide which is susceptible to tryptic digestion. Interestingly, over the first half hour after exposure to trypsin, the activity of the enzyme increased (very slightly), which may be an indication that biodegradation of the protective shell around the enzyme is occurring and thereby increasing the activity of the enzyme. This increase, however, is minor and is followed by a more rapid decrease of activity over the next four hours, indicative of ASNase degradation. Shen and co-workers72,93 have evaluated the stability of ASNase modified with silk fibroin and sericin and have also concurrently observed only a decrease of enzymatic activity over time, indicative that the conjugated proteins cannot be selectively degraded without compromising ASNase activity.In contrast to polypeptides, polysaccharides may be degraded under conditions which, in principle, should not affect the enzyme. Marshall and Rabinowitz81 have evaluated the biodegradation of a trypsin–dextran conjugate in the presence of dextranase. The authors interestingly observed no change in esterolytic or caseinolytic activity following treatment with the enzyme. In addition, the authors observed that the susceptibility of the conjugates to inhibition with ovomucoid increased following dextranase treatment, indicating that removal of dextran increased the accessibility of the inhibitor to the active site of the conjugated enzyme. Consequently, the authors concluded that the non-recovery of activity of the conjugate following dextranase treatment is due to carbohydrate attachment per se rather than concomitant burying of enzyme's active site by polymer chains. More recently, Duncan et al.79 have evaluated the biodegradability of trypsin–dextrin conjugates by α-amylase. The enzymatic activity of the conjugate increased upon exposure to the enzyme, though the extent of regeneration was difficult to interpret. The authors indicated that this would be related to the rate of polymer degradation, the liberation of active protein with little or no glucose oligomers attached, and maintenance of protein integrity during conjugation and subsequent biodegradation.
6.2. Labile linkers for non-biodegradable polymers
A relatively recent approach for regenerating enzymatic activity of protein–polymer conjugates in the body is to introduce a cleavable linker between the polymer and the enzyme.109 Examples of conjugates of lysozyme with MPEG containing a labile linker are found in Table 4. Lysozyme is an excellent model protein to evaluate this phenomenon because conjugation of a single 5 kDa MPEG chain to a lysine residue on this protein has been shown to completely eliminate enzymatic activity.60,61,110,111 These linkers are intercalated between the polymer and the protein and are designed to release these two moieties by hydrolysis in the body. This approach is, in principle, amenable to any non-biodegradable polymer. These authors have exploited cleavable linkers to reactivate the enzyme, which in most cases reached ∼100% of the activity of the native enzyme after an appropriate time of incubation. One exception is the conjugate prepared by Roberts and Harris,110 which only achieved 60% reactivation. This may be due to the fact that in this case linker cleavage is not “traceless” in as much as the lysine residue is not recovered but remained modified (with a small molecule) following linker cleavage. This has been shown in a study by Zhao et al.61 who have modified the amino groups on lysozyme with small molecules such as maleic anhydride, resulting in a reduction of activity of 60% and thereby demonstrating that the attachment of a small molecule to an amine of lysozyme can also inhibit activity. The authors further speculated that possible denaturation and conformational changes in heavily PEGylated molecules, which are not reversible upon release of the polymer contribute to this phenomenon.6.3. Side-chain biodegradable polymers
Vinyl polymers are generally considered to be non-biodegradable along their main-chain. One exception, though not treated herein, are poly(alkyl cyanoacrylate)s. However, the functional side-chains of main-chain non-biodegradable polymers are generally attached to the backbone by ester or amide bonds, which have the possibility of degrading inside the body. On the one hand, this process is undesirable because of complications arising from the need to evaluate the potential bioactivity of the biodegradation products. On the other, this process can, over time, dramatically reduce the molecular weight of the polymer, especially when the side-groups have large molecular weights. This holds particularly true for comb-type polymers which possess large side-chains such as those employed by Qian et al.89 and Lele et al.53 (5 and 1.1 kDa, respectively). Clearly, the amide and ester groups present on typical (meth)acrylate or (meth)acrylamide polymers are expected to be quite stable during the life span of the conjugate and thus should not affect the activity of the conjugate over this period. In terms of ultimate fate however, there is unfortunately very little information available on the rates of non-specific hydrolysis of polymer side-chains in the body (or in simulated conditions) or on the biological fate in vivo of many common polymers, whether biodegradable or not.112–119 Further studies will become necessary as future research in this field is pursued.6.4. General trends
In general, in the examples cited, polypeptide biodegradation resulted in little or no recovery of enzyme activity, which may result from the similarity between polypeptide and enzyme degradation conditions. The selective biodegradation of polysaccharides, however, is possible and lead in certain cases to increased accessibility of small-molecules to the active site of the enzyme, though this did not always correlate with a complete recovery of activity. This phenomenon was also observed for when certain “non-traceless” cleavable linkers were used to prepare biodegradable MPEG–lysozyme conjugates.7. Outlook
Through this review article we've highlighted trends observed using new protein–polymer conjugation strategies, with particular emphasis on how they relate to enzymatic activity. It should become apparent that modern tools for preparing protein–polymer conjugates allow for exceptional freedom in conjugate design and as such it is somewhat difficult to find comparable conjugates within the literature. In addition, there are very few systematic studies which have been performed to assess the individual contributions of many of the factors discussed herein, which makes it very difficult to make general statements. Owing to the specificity and dissimilarity of trends observed and summarized in Table 7, we feel it would be premature to provide general guidelines regarding optimum molecular weight, residue for modification, distance from active sites, etc., for maintaining catalytic activity of conjugates. We have however provided a summary of the specific effect of state-of-the-art protein modifications on activity and have summarized trends observed for each protein in Table 7.As more and more conjugates are prepared in the coming years, the generality of the statements made herein, for select proteins, may be verified or negated. One clear point which is to be made with this review is that no one factor is uniquely responsible for conjugate properties, rather the cumulative effect of the applicable factors described herein governs its ultimate properties. It is our hope with this contribution to summarize the state-of-the-art in this rapidly expanding field with the desire to provide (at least tentative) guidelines for preparing future functional protein–polymer constructs.
One final topic which we have not touched upon in this contribution is that of immunogenicity. Immunogenicity is an important and intrinsic property of the polymer and/or the conjugate and its evaluation must be made concurrently with the development of such systems, independently of the polymer used. Even PEG, with the plethora of data to support its use for therapeutic purposes, cannot escape this requirement especially as recent studies are beginning to identify anti-PEG antibodies in animals.120–122
8. List of abbreviations for tables
| AADG | acrylamido-2-deoxy-D-glucose |
| ABr | via acid bromide |
| Alk | alkylation with α-bromoester |
| AzoT | via azo trypsin |
| bisAlk | bis-alkylation via a three-carbon bridge |
| BMA | n-butyl methacrylate |
| CAP | cellulose acetate phthalate |
| CDI | via carbodiimide |
| CMC | carboxymethyl cellulose |
| CM-PβCD | O-carboxymethyl-poly-β-cyclodextrin |
| CNBr | cyanogen bromide |
| Eudragit-S | 1:2 methacrylic acid-co-methyl methacrylate |
| HP-55 | hydroxy propylmethylcellulose phthalate |
| Hyd | via hydrazide |
| GA | via gluteraldehyde |
| GEMA | glucosyoxylethyl methacrylate |
| MA | maleic anhydride |
| MAA | methacrylic acid |
| Mal | via Michael addition with maleimide |
| MMA | methyl methacrylate |
| MPEG | α-methoxy-poly(ethylene glycol) |
| MPEG-deg | MPEG with degradable linker |
| MPEG2 | double branched MPEG |
| MPM-06 | methylacrylate-methacrylic acid-methylmethacrylate copolymer |
| n.c. | not characterized |
| NC | non-covalent |
| NCA | via α-amino acid N-carboxy anhydride polymerization |
| n.g. | not given |
| NHS | via N-hydroxysuccinimide active ester |
| NIPAAm | N-isopropylacrylamide |
| N,O-CM-Chit | N,O-CM-chitosan |
| NPC | p-nitrophenylcarbonate |
| OTs | via tosylate |
| PAGM | poly(aminoglucose methacrylate) |
| PAsp | poly(aspartic acid) |
| PCVBPC | poly(3-carbamoyl-1-(p-vinylbenzyl)pyridinium chloride) |
| PDMAEMA | poly(N,N-dimethylaminoethyl methacrylate) |
| PDMAPS | poly(3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) |
| PEG | poly(ethylene glycol) |
| PEO | poly(ethylene oxide) |
| PHPMA | poly(2-hydroxypropyl methacrylamide) |
| PM13 | copolymer of poly(oxyethylene) allyl methyl diether and maleic anhydride |
| PM100 | copolymer of poly(oxyethylene) 2-methyl-2-propenyl methyl diether and maleic anhydride |
| PMPC | poly(2-methacryloyloxyethyl phosphorylcholine) |
| PNaPS | poly(sodium 4-styrenesulfonate) |
| PNIPAAm | poly(N-isopropylacrylamide) |
| pNP | via p-nitrophenyl ester |
| POZ | poly(2-ethyl-oxazoline) |
| PPEGA | poly(polyethylene glycol monomethyl ether acrylate) |
| PPEGMA and PPEGMA475 | poly(polyethylene glycol monomethyl ether methacrylate) |
| PSucr | polymeric sucrose |
| PVP | poly(vinyl pyrrolidone) |
| RA | via reductive alkylation |
| RPE and REP | block copolymers of ethylene oxide and propylene oxide, (R: radical, P: propylene oxide, E: ethylene oxide) |
| SC | via succinimidyl carbonate |
| SS | via disulfide exchange |
| TCT | via 2,4,6-trichloro[1,3,5]-triazine |
| TGase | transglutaminase |
| TT | via thiazolidine-2-thione coupling |
| VP | vinyl pyrrolidone |
| VS | via vinyl sulfone. |
Acknowledgements
H.-A.K. thanks the Swiss National Science Foundation, the NCCR Nanoscale Science, and the European Commission for supporting his laboratory's activities on peptide/protein–synthetic polymer conjugates. M.A.G. gratefully acknowledges a scholarship from the Fonds Québecois de la Recherche sur la Nature et les Technologies (Quebec, Canada) for funding his postdoctoral research on protein–polymer conjugates with H.-A. K from 2007–2009.Notes and references
- E. Pedone and S. Brocchini, React. Funct. Polym., 2006, 66, 167–176 CrossRef CAS.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- G. Pasut, M. Sergi and F. M. Veronese, Adv. Drug Delivery Rev., 2008, 60, 69–78 CrossRef CAS.
- J. M. Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539–551 CrossRef CAS.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS.
- S. Brocchini, A. Godwin, S. Balan, J.-w. Choi, M. Zloh and S. Shaunak, Adv. Drug Delivery Rev., 2008, 60, 3–12 CrossRef CAS.
- A. J. de Graaf, M. Kooijman, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2009, 20, 1281–1295 CrossRef CAS.
- H. B. F. Dixon, J. Protein Chem., 1984, 3, 99–108 CAS.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243–254 CrossRef CAS.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253–262 CrossRef CAS.
- D. R. W. Hodgson and J. M. Sanderson, Chem. Soc. Rev., 2004, 33, 422–430 RSC.
- R. E. Connor and D. A. Tirrell, J. Macromol. Sci., Polym. Rev., 2007, 47, 9–28 CAS.
- A. J. Link, M. L. Mock and D. A. Tirrell, Curr. Opin. Biotechnol., 2003, 14, 603–609 CrossRef CAS.
- J. C. M. van Hest, J. Macromol. Sci., Polym. Rev., 2007, 47, 63–92 CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- H. G. Börner and H. Schlaad, Soft Matter, 2007, 3, 394–408 RSC.
- D. W. P. M. Löwik, L. Ayres, J. M. Smeenk and J. C. M. van Hest, Adv. Polym. Sci., 2006, 202, 19–52.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M.v. Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- A. Hirao and M. Hayashi, Acta Polym., 1999, 50, 219–231 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1 10.1039/b9py00363k.
- Y. Inada, M. Furukawa, H. Sasaki, Y. Kodera, M. Hiroto, H. Nishimura and A. Matsushima, Trends Biotechnol., 1995, 13, 86–91 CrossRef CAS.
- V. Gaberc-Porekar, I. Zore, B. Podobnik and V. Menart, Curr. Opin. Drug Discovery Dev., 2008, 11, 242–250 CAS.
- A. Fontana, B. Spolaore, A. Mero and F. M. Veronese, Adv. Drug Delivery Rev., 2008, 60, 13–28 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25, 247–249 CrossRef CAS.
- Y. Ashihara, T. Kono, S. Yamazaki and Y. Inada, Biochem. Biophys. Res. Commun., 1978, 83, 385–391 CrossRef CAS.
- A. Matsushima, H. Nishimura, Y. Ashihara and Y. Inada, Chem. Lett., 1980, 773–776 CrossRef CAS.
- J.-F. Zhang, L.-Y. Shi and D.-Z. Wei, Biotechnol. Lett., 2004, 26, 753–756 CrossRef CAS.
- A. L. Soares, G. M. Guimarães, B. Polakiewicz, R. N. d. M. Pitombo and J. Abrahão-Neto, Int. J. Pharm., 2002, 237, 163–170 CrossRef CAS.
- C. Monfardini, O. Schiavon, P. Caliceti, M. Morpurgo, J. M. Harris and F. M. Veronese, Bioconjugate Chem., 1995, 6, 62–69 CrossRef CAS.
- F. M. Veronese, C. Monfardini, P. Caliceti, O. Schiavon, M. D. Scrawen and D. Beer, J. Controlled Release, 1996, 40, 199–209 CrossRef CAS.
- N. Verma, K. Kumar, G. Kaur and S. Anand, Crit. Rev. Biotechnol., 2007, 27, 45–62 CrossRef CAS.
- J. A. Rodríguez-Martínez, I. Rivera-Rivera, R. J. Solá and K. Griebenow, Biotechnol. Lett., 2009, 31, 883–887 CrossRef CAS.
- I. J. Castellanos, W. Al-Azzam and K. Griebenow, J. Pharm. Sci., 2005, 94, 327–340 CrossRef CAS.
- H.-C. Chiu, S. Zalipsky, P. Kopečková and J. Kopeček, Bioconjugate Chem., 1993, 4, 290–295 CrossRef CAS.
- H. F. Gaertner and A. J. Puigserver, Enzyme Microb. Technol., 1992, 14, 150–155 CrossRef CAS.
- L. Goldstein, Biochemistry, 1972, 11, 4072–4084 CrossRef CAS.
- A. Abuchowski and F. F. Davis, Biochim. Biophys. Acta, Protein Struct., 1979, 578, 41–46 CrossRef CAS.
- B. Treetharnmathurot, C. Ovartlarnporn, J. Wungsintaweekul, R. Duncan and R. Wiwattanapatapee, Int. J. Pharm., 2008, 357, 252–259 CrossRef CAS.
- T. So, T. Ueda, Y. Abe, T. Nakamata and T. Imoto, J. Biochem., 1996, 119, 1086–1093 CAS.
- J. R. Uren and R. C. Ragin, Cancer Res., 1979, 39, 1927–1933 CAS.
- T. Yoshimura, A. Imanishi and T. Isemura, J. Biochem., 1968, 63, 730–737 CAS.
- Z.-R. Lu, P. Kopečková, Z. Wu and J. Kopeček, Bioconjugate Chem., 1998, 9, 793–804 CrossRef CAS.
- D. Oupický, K. Ulbrich and B. Říhová, J. Bioact. Compat. Polym., 1999, 14, 213–231 CAS.
- Z. Ding, G. Chen and A. S. Hoffman, J. Biomed. Mater. Res., 1998, 39, 498–505 CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- D. Miyamoto, J. Watanabe and K. Ishihara, Biomaterials, 2004, 25, 71–76 CrossRef CAS.
- R. J. Solá and K. Griebenow, FEBS J., 2006, 273, 5303–5319 CrossRef CAS.
- S. D. Varfolomeev and K. G. Gurevich, Russ. Chem. Bull., 2001, 50, 1709–1717 Search PubMed.
- G. J. Palm, J. Lubkowski, C. Derst, S. Schleper, K.-H. Röhm and A. Wlodawer, FEBS Lett., 1996, 390, 211–216 CrossRef CAS.
- S. Balan, J.-w. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–76 CrossRef CAS.
- T. Higashi, F. Hirayama, S. Yamashita, S. Misumi, H. Arima and K. Uekama, Int. J. Pharm., 2009, 374, 26–32 CrossRef CAS.
- R. B. Greenwald, K. Yang, H. Zhao, C. D. Conover, S. Lee and D. Filpula, Bioconjugate Chem., 2003, 14, 395–403 CrossRef CAS.
- H. Zhao, K. Yang, A. Martinez, A. Basu, R. Chintala, H.-C. Liu, A. Janjua, M. Wang and D. Filpula, Bioconjugate Chem., 2006, 17, 341–351 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS.
- J.-H. Seo, R. Matsuno, Y. Lee, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 4859–4867 CrossRef CAS.
- J. Córdova, J. D. Ryan, B. B. Boonyaratanakornkit and D. S. Clark, Enzyme Microb. Technol., 2008, 42, 278–283 CrossRef CAS.
- Y. Ito, M. Kotoura, D.-J. Chung and Y. Imanishi, Bioconjugate Chem., 1993, 4, 358–361 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- V. V. Mozhaev, N. S. Melik-Nubarov, M. V. Sergeeva, V. Šikšnis and K. Martinek, Biocatal. Biotransform., 1990, 3, 179–187 CrossRef CAS.
- T. Yoshimoto, K. Takahashi, A. Ajima, A. Matsushima, Y. Saito, Y. Tamaura and Y. Inada, FEBS Lett., 1985, 183, 170–172 CrossRef CAS.
- Y.-Q. Zhang, W.-L. Zhou, W.-D. Shen, Y.-H. Chen, X.-M. Zha, K. Shirai and K. Kiguchi, J. Biotechnol., 2005, 120, 315–326 CrossRef CAS.
- I. Vīna, A. Karsakevich and M. Bekers, J. Mol. Catal. B: Enzym., 2001, 11, 551–558 CrossRef CAS.
- Y. Kodera, T. Sekine, T. Yasukohchi, Y. Kiriu, M. Hiroto, A. Matsushima and Y. Inada, Bioconjugate Chem., 1994, 5, 283–286 CrossRef CAS.
- H. K. Kim and T. G. Park, Enzyme Microb. Technol., 1999, 25, 31–37 CrossRef CAS.
- P. Wang, T. G. Hill, C. A. Wartchow, M. E. Huston, L. M. Oehler, M. B. Smith, M. D. Bednarski and M. R. Callstrom, J. Am. Chem. Soc., 1992, 114, 378–380 CrossRef CAS.
- A. Lääne, V. Chytrý, M. Haga, P. Sikk, A. Aaviksaar and J. Kopeček, Collect. Czech. Chem. Commun., 1981, 46, 1466–1473 CAS.
- P. V. Sundaram and R. Venkatesh, Protein Eng., Des. Sel., 1998, 11, 699–705 CrossRef CAS.
- R. Duncan, H. R. P. Gilbert, R. J. Carbajo and M. J. Vicent, Biomacromolecules, 2008, 9, 1146–1154 CrossRef CAS.
- B. Treetharnmathurot, L. Dieudonné, E. L. Ferguson, D. Schmaljohann, R. Duncan and R. Wiwattanapatapee, Int. J. Pharm., 2009, 373, 68–76 CrossRef CAS.
- J. J. Marshall and M. L. Rabinowitz, J. Biol. Chem., 1976, 251, 1081–1087 CAS.
- M. L. Villalonga, R. Villalonga, L. Mariniello, L. Gómez, P. Di Pierro and R. Porta, World J. Microbiol. Biotechnol., 2006, 22, 595–602 CrossRef CAS.
- N. Rajalakshmi and P. V. Sundaram, Protein Eng., Des. Sel., 1995, 8, 1039–1047 CrossRef CAS.
- D. Venturoli and B. Rippe, Am. J. Physiol.: Renal Physiol., 2005, 288, F605–613 CAS.
- S. S. Stivala, J. Kimura and L. Reich, Thermochim. Acta, 1981, 50, 111–122 CrossRef CAS.
- T. C. Le, B. D. Todd, P. J. Daivis and A. Uhlherr, J. Chem. Phys., 2009, 131, 044902–044910 CrossRef CAS.
- M. J. Poznansky, M. Shandling, M. A. Salkie, J. Elliott and E. Lau, Cancer Res., 1982, 42, 1020–1025 CAS.
- Y.-J. Sheng, K.-L. Cheng and C.-C. Ho, J. Chem. Phys., 2004, 121, 1962–1968 CrossRef CAS.
- G. Qian, J. Ma, J. Zhou, B. He and D. Wang, Polym. Adv. Technol., 1997, 8, 581–586 CrossRef CAS.
- G. Qian, J. Ma, J. Zhou and B. He, React. Funct. Polym., 1997, 32, 117–121 CrossRef CAS.
- Y. Kamisaki, H. Wada, T. Yagura, A. Matsushima and Y. Inada, J. Pharmacol. Exp. Ther., 1981, 216, 410–414 CAS.
- H. Sasaki, Y. Ohtake, A. Matsushima, M. Hiroto, Y. Kodera and Y. Inada, Biochem. Biophys. Res. Commun., 1993, 197, 287–291 CrossRef CAS.
- Y.-Q. Zhang, M.-L. Tao, W.-D. Shen, J.-P. Mao and Y.-h. Chen, J. Chem. Technol. Biotechnol., 2006, 81, 136–145 CrossRef CAS.
- G. Qian, J. Zhou, J. Ma, D. Wang and B. He, Artif. Cells, Blood Substitutes, Biotechnol., 1996, 24, 567–577 Search PubMed.
- M. Yan, J. Ge, W. Dong, Z. Liu and P. Ouyang, Biochem. Eng. J., 2006, 30, 48–54 CrossRef CAS.
- D. Miyamoto, J. Watanabe and K. Ishihara, J. Appl. Polym. Sci., 2004, 91, 827–832 CrossRef CAS.
- M. Fujimura, T. Mori and T. Tosa, Biotechnol. Bioeng., 1987, 29, 747–752 CrossRef CAS.
- M. Fernández, M. L. Villalonga, A. Fragoso, R. Cao, M. Baños and R. Villalonga, Process Biochem., 2004, 39, 535–539 CrossRef CAS.
- R. Villalonga, M. Fernández, A. Fragoso, R. Cao, L. Mariniello and R. Porta, Biotechnol. Appl. Biochem., 2003, 38, 53–59 CrossRef CAS.
- M. Fernández, M. L. Villalonga, A. Fragoso, R. Cao and R. Villalonga, Biotechnol. Appl. Biochem., 2002, 36, 235–239 CrossRef CAS.
- M. Fernández, A. Fragoso, R. Cao and R. Villalonga, Process Biochem., 2005, 40, 2091–2094 CrossRef CAS.
- M. Fernández, M. L. Villalonga, J. Caballero, A. Fragoso, R. Cao and R. Villalonga, Biotechnol. Bioeng., 2003, 83, 743–747 CrossRef CAS.
- B.-U. von Specht and W. Brendel, Biochim. Biophys. Acta, Enzymol., 1977, 484, 109–114 Search PubMed.
- H. Lee and T. G. Park, Biotechnol. Prog., 1998, 14, 508–516 CrossRef CAS.
- X. Yuan, Y. Yamasaki, A. Harada and K. Kataoka, Polymer, 2005, 46, 7749–7758 CrossRef CAS.
- M. Danial, H.-A. Klok, W. Norde and M. A. Cohen Stuart, Langmuir, 2007, 23, 8003–8009 CrossRef CAS.
- D. Miyamoto, J. Watanabe and K. Ishihara, J. Appl. Polym. Sci., 2005, 95, 615–622 CrossRef CAS.
- W. R. Gombotz and D. K. Pettit, Bioconjugate Chem., 1995, 6, 332–351 CrossRef CAS.
- D. Filpula and H. Zhao, Adv. Drug Delivery Rev., 2008, 60, 29–49 CrossRef CAS.
- M. J. Roberts and J. M. Harris, J. Pharm. Sci., 1998, 87, 1440–1445 CrossRef CAS.
- S. Lee, R. B. Greenwald, J. McGuire, K. Yang and C. Shi, Bioconjugate Chem., 2001, 12, 163–169 CrossRef CAS.
- N. Bertrand, J. G. Fleischer, K. M. Wasan and J.-C. Leroux, Biomaterials, 2009, 30, 2598–2605 CrossRef CAS.
- Y. Murakami, Y. Tabata and Y. Ikada, Drug Delivery, 1997, 4, 23–31 CrossRef CAS.
- Y. Takakura, T. Fujita, M. Hashida and H. Sezaki, Pharm. Res., 1990, 7, 339–346 CrossRef CAS.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Pharmacol., 1995, 47, 479–486 CAS.
- L. W. Seymour, R. Duncan, J. Strohalm and J. Kopeček, J. Biomed. Mater. Res., 1987, 21, 1341–1358 CAS.
- L. W. Seymour, Y. Miyamoto, H. Maeda, M. Brereton, J. Strohalm, K. Ulbrich and R. Duncan, Eur. J. Cancer, 1995, 31, 766–770 CrossRef.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1995, 84, 349–354 CrossRef CAS.
- X. Y. Wang, T. Ishida and H. Kiwada, J. Controlled Release, 2007, 119, 236–244 CrossRef CAS.
- A. Judge, K. McClintock, J. R. Phelps and I. MacLachlan, Mol. Ther., 2006, 13, 328–337 CrossRef CAS.
- K. Środa, J. Rydlewski, M. Langner, A. Kozubek, M. Grzybek and A. F. Sikorski, Cell. Mol. Biol. Lett., 2005, 10, 37–47 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |