Encapsulation of the uranyl dication†
Stephan
Beer‡
,
Orion B.
Berryman‡
,
Dariush
Ajami
and
Julius
Rebek Jr.
*
Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: jrebek@scripps.edu
First published on 21st May 2010
Abstract
We report here the application of self-assembly to the sequestration of uranyl ion, the dominant form of uranium on Earth. The assembly presents the ion with convergent carboxylate ligands, isolates it from solvent, encapsulates it with aromatic panels and allows its extraction from aqueous solutions in the presence of excess brine.
Introduction
Reversible encapsulation complexes are systems in which molecular guests are more or less surrounded by a self-assembled host. Weak, intermolecular forces hold the assemblies together: hydrogen bonds;1–7 hydrophobic effects;8 salt bridges and metal/ligand interactions.9,10 In the latter, the metal centers act as hinges at the corners of the capsule assembling aromatic panels that create a cavity. We introduce here the use of a metal ion to gather aromatic panels and encapsulate itself at the center. This alternative is applied to the molecular recognition of uranyl ion.The highly oxidizing conditions on Earth ensure that virtually all aqueous uranium is U(VI) in the form of uranyl ion (UO22+). Several methods have been developed for the recovery of uranium from wet-process phosphonic acids.11 However, sequestering this ion from dilute solutions such as sea water continues to remain a challenge because of the uranyl ion's distinctive shape (Fig. 1, left). The linear uranyl dication—which prefers planar coordination about its equator—prevents the use of conventional chelating ligands, such as ethylenediaminetetraacetic acid that effectively target spherical cations.12
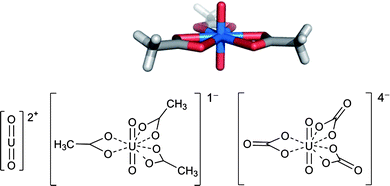 | ||
| Fig. 1 Linear structure of the uranyl dication (left) and its crystalline complex with acetate (top and bottom middle) and ChemDraw of the carbonate monomer (bottom right). | ||
Typically, the oxygens of the uranyl ion are chemically inert, and there are generally 5 or 6 additional donor atoms around the metal. UO22+ is a hard Lewis acid and prefers to interact with hard oxygen donors; accordingly, many carboxylate-containing ligands are known for the uranyl ion. A crystallographic example of planar hexacoordinate uranyl triacetate13 is shown in Fig. 1. Uranyl tricarbonate is the predicted form of uranium in sea water and is recognized to exist as the monomer [UO2(CO3)3]4− (Fig. 1, bottom right) or the trimer [UO2(CO3)2]36− in highly ionic solutions.14 The carboxylates act as bidentate ligands with an appropriate “bite” angle that allows three carboxylates to fit around one metal center with optimal positioning of the oxygen atoms.
The synthesis of new ligands that effectively bind UO22+ continues to attract interest. Raymond15 and co-workers have comprehensively analyzed the requirements for the molecular recognition of uranyl ion and have synthesized a series of tricarboxylate ligands that feature the ideal stereoelectronics for chelation. The stereoelectronic effects at carboxyl oxygens are well known16,17 and are reflected in many enzyme crystal structures.18 Raymond's ligands are shown in Fig. 2 and present carboxylate oxygens in optimal approaches to the metal center. In addition, these designs also offer a complementary hydrogen bond donor to the uranyl oxygen but the single bonds feature several degrees of rotational freedom that pose entropic penalties to chelation.19 Accordingly, effective chelating agents—those that might be used for sequestration of uranyl from dilute aqueous solutions—are lacking.
 | ||
| Fig. 2 Tripodal ligands that bind uranyl ion with ammonium sites for uranyl oxygen recognition. | ||
Encapsulation in apolar environments is known to amplify electrostatic interactions,20 stabilize contact ion pairs21 and enhance other forces such as hydrogen bonds.22 These effects result from the lower dielectric constant of the hydrophobic environment through the exclusion of solvent water molecules—much as is the case in enzyme interiors.23 Our focus was on encapsulating the ion, with the premise that the attraction between the uranium ion and carboxylate ligands would be enhanced when isolated in a hydrophobic environment.
Results and discussion
To encapsulate the uranyl ion we chose 2,6-terphenyl carboxylic acids as the target subunits for assembly. Typically, metals prefer one or two 2,6-terphenyl carboxylic acid ligands around their coordination sphere. Due to the large size and planar coordination of the uranyl ion, three 2,6-terphenyl carboxylic acid ligands are of appropriate size and shape to surround the uranyl ion with aromatic panels and present it with carboxylates that satisfy the planar coordination geometry on the metal ion (Fig. 3). This molecular arrangement ensures that the more basic lone pairs of the carboxylate engage the metal center, while the flanking phenyl groups provide a physical barrier to solvent (water) molecules.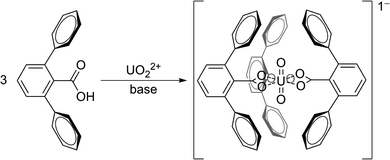 | ||
| Fig. 3 General schematic of 2,6-terphenyl carboxylic acids binding to uranyl ion. | ||
The 2,6-terphenyl carboxylic acids 1 and 2 are readily obtained from an elegant synthesis developed by Hart et al. (Fig. 4).24 The yield is high and tolerant of many substituents on the central and flanking phenyl groups.25 The known phenyl substituted 2,6-terphenyl ligand 1 and methoxy derivative 2 were synthesized by aryl cross couplings of 2,6-dichloroiodobenzene and the appropriate aryl Grignard reagents. Ancillary functionality could be incorporated into the 2,6-terphenyl ligand by deprotecting the methoxy substituent of 2 with BBr3 in CH2Cl2 at −78 °C (Fig. 4). Novel hydroxy substituted ligand 3 is isolated in 62% yield as a white powder. Other demethylation procedures gave side products including cyclization reactions and decarboxylation.
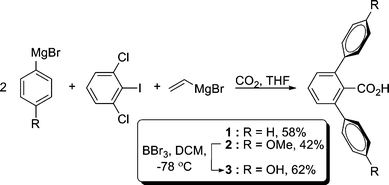 | ||
| Fig. 4 Synthesis of 2,6-terphenyl carboxylic acid ligands 1–3. | ||
In preliminary experiments, mixing methanol solutions of uranyl nitrate UO2(NO3)2 or diacetate UO2(OAc)2 with the triethylammonium salt of 1 (Et3N+1−) resulted in an immediate light yellow precipitate of the corresponding uranium complex [Et3NH]+[UO2(1)3]−. Slow evaporation of concentrated dichloromethane solutions of this precipitate gave pale yellow single crystals suitable for X-ray diffraction.§ The uranium complex crystallizes in the space group P21/n and the structure is shown in Fig. 5.26 Three 2,6-terphenyl ligands serve to bind to the uranyl ion as well as enclose the metal center in hydrophobic aromatic rings. Edge-to-face contacts exist between the peripheral aromatic rings with a closest hydrogen to arene-plane distance of approximately 3.18 Å. These edge-to-face interactions27 likely play a role in the kinetic stability of this complex toward ligand substitution. This is a unique crystalline example of three bidentate 2,6-terphenyl carboxylic acids surrounding one metal center. Interestingly, the triethylammonium cation is encapsulated by two adjacent UO2(1)3− ions and this arrangement propagates throughout the crystal producing 1-dimensional chains (Fig. 6).
![Crystal structure of [Et3N]+[UO2(1)3]− highlighting the three 2,6-terphenyl ligands (left) and the encapsulated UO2 ion (right). Carbon is gray, oxygen red, uranium blue and hydrogens white. Solvent, some hydrogens and counter cations have been removed for clarity.](/image/article/2010/SC/c0sc00116c/c0sc00116c-f5.gif) | ||
| Fig. 5 Crystal structure of [Et3N]+[UO2(1)3]− highlighting the three 2,6-terphenyl ligands (left) and the encapsulated UO2 ion (right). Carbon is gray, oxygen red, uranium blue and hydrogens white. Solvent, some hydrogens and counter cations have been removed for clarity. | ||
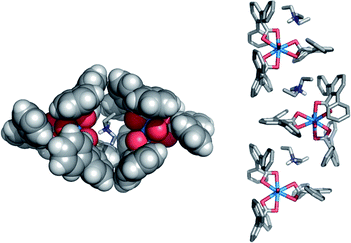 | ||
| Fig. 6 Illustration of the enclosed environment around the Et3NH cation (stick, left) and the 1-D chains formed in the crystalline phase (right). | ||
To the best of our knowledge, this is the only crystal structure example of a 2,6-terphenyl carboxylic acid coordination complex that does not contain iron.28 A search of the Cambridge Structural Database29 (CSD) for all 2,6-terphenyl carboxylate molecules bound to any atom revealed 69 structures. Surprisingly, all of the 69 structures contained 2,6-terphenyl carboxylates bound to at least one Fe atom (one structure contained both thallium and iron). A large majority of the hits (51) contained 2,6-terphenyl ligands both bridging two metals and bound to one metal center in the same structure; 14 structures contained two ligands around a single Fe atom while only two structures included one ligand around a single Fe atom. An example was observed for one ligand bridging two Fe atoms. There appears to be only one example in the CSD of three 2,6-terphenyl acids around a single metal center (not all the acids were bound to the Fe through both oxygens). The structures presented here are unique because the large size and planar coordination of the uranyl ion allows for three 2,6-terphenyl carboxylate ligands to bind in a bidentate fashion to one uranium.
The Earth's oceans contain uranium at a concentration of around three parts per billion. Generous quantities of uranium could be obtained if this valuable element could be sequestered from the copious amount of sea water on Earth. However, the low concentration of uranium and presence of other salts makes this an inherently challenging enterprise. We were interested in using the 2,6-terphenyl carboxylic ligands to extract uranium from aqueous sources and found that the same encapsulation complex is obtained from liquid/liquid extractions. For examples, aqueous solutions (0.017 M) of uranyl nitrate buffered to pH 5 with acetate—or containing a 10-fold excess of NaCl—were extracted with 1 in an organic solvent (CDCl3). 1H NMR spectroscopy confirmed that the uranium was extracted as the complex [Et3NH]+[UO2(1)3]− and ICP analysis of the aqueous phase showed that 74% of the uranium is extracted into the organic layer in the presence of NaCl. The uranium can be fully recovered by stirring with dilute HNO3 (see ESI†).
The uranyl binding capability of the 2,6-terphenyl acid class appears to be general. Analogous results were obtained for the methoxy substituted 2 and hydroxy derivative 3. Three such ligands assemble around one uranyl ion and these complexes have been characterized by 1H, 13C NMR, elemental analysis, and mass spectrometry. One notable difference is the solubility of the complex [iPr2EtNH]+[UO2(3)3]− obtained when the hydroxy substituted ligand 3 is treated with UO2(NO3)2 in the presence of the hindered Hünig's base (iPr2EtN).
In contrast to the other complexes, [iPr2EtNH]+[UO2(3)3]− does not precipitate from methanol. Crystals of this complex suitable for X-ray diffraction were obtained by evaporating the solvent followed by extraction with CHCl3 and cooling this solution to 0 °C. The [iPr2EtNH]+[UO2(3)3]− complex crystallizes in the Cc space group with 4 molecules per unit cell (Fig. 7).§ The ligand arrangement around the uranyl ion is structurally similar to the complex formed between UO2 and 1 with differences observed in the packing arrangement and lattice solvent.30 The U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is typically inert.31 However, studies suggest that the effective charge on U can be closer to 3+, resulting in UO2 oxygens that are Lewis basic15,19,32 and a number of cations33 and a Lewis acid34 have been observed interacting with the uranyl oxygen. In this crystal, two water molecules seal each end of the capsule through hydrogen bonding to the phenolic OHs but do not engage the uranyl ion's oxygens, highlighting the isolating role of the hydrophobic phenyl substituents.
O bond is typically inert.31 However, studies suggest that the effective charge on U can be closer to 3+, resulting in UO2 oxygens that are Lewis basic15,19,32 and a number of cations33 and a Lewis acid34 have been observed interacting with the uranyl oxygen. In this crystal, two water molecules seal each end of the capsule through hydrogen bonding to the phenolic OHs but do not engage the uranyl ion's oxygens, highlighting the isolating role of the hydrophobic phenyl substituents.
![Stick (left) and CPK (right) representation of the [iPr2EtNH]+[UO2(3)3]− crystal structure highlighting the peripheral phenol interaction with water molecules. Water oxygens labeled Ow; hydrogens and cations have been removed for clarity.](/image/article/2010/SC/c0sc00116c/c0sc00116c-f7.gif) | ||
| Fig. 7 Stick (left) and CPK (right) representation of the [iPr2EtNH]+[UO2(3)3]− crystal structure highlighting the peripheral phenol interaction with water molecules. Water oxygens labeled Ow; hydrogens and cations have been removed for clarity. | ||
The kinetic stability of complexes formed between UO22+ and 2,6-terphenyl acid ligands 1–3 was established by 1H NMR experiments. The interactions of carboxylates with the uranyl ion are typically fast exchange processes, but for the complexes of 1–3 they are relatively slow. Separate signals are seen in the 1H NMR spectrum of [Et3NH]+[UO2(1)3]− and excess Et3N+1− in CD2Cl2 solvent (Fig. 8).35 In the hydrophobic environment of the capsule, the charge–charge interactions and edge-to-face attractions are amplified.36 The high energetic barrier to the exchange of ligands suggests tight binding and large association constants for the capsular assembly, even though 4 molecules must be brought together to form the complex. The phenolic OH groups of 3 suggest a means to connect the subunits, as in Raymond's compounds, into a single chelating agent that overcomes these entropic costs and allows a more straightforward and quantitative measure of the appropriate association constants.
![1H NMR spectra illustrating the high kinetic stability of [Et3NH]+[UO2(1)3]− in CD2Cl2 solution; (A) [Et3NH]+[UO2(1)3]−; (B) 1 eq. Et3N+1− added; (C) 5 total eq. Et3N+1− added; (D) 10 total eq. Et3N+1− added. The [Et3NH]+[UO2(1)3]− signals are marked by blue squares and unbound Et3N+1− is marked by red circles.](/image/article/2010/SC/c0sc00116c/c0sc00116c-f8.gif) | ||
| Fig. 8 1H NMR spectra illustrating the high kinetic stability of [Et3NH]+[UO2(1)3]− in CD2Cl2 solution; (A) [Et3NH]+[UO2(1)3]−; (B) 1 eq. Et3N+1− added; (C) 5 total eq. Et3N+1− added; (D) 10 total eq. Et3N+1− added. The [Et3NH]+[UO2(1)3]− signals are marked by blue squares and unbound Et3N+1− is marked by red circles. | ||
Summary
Reversibly assembled capsules have been extensively developed for anionic, cationic and neutral guest species and the aromatic panels that define the space provide a hydrophobic cavity within the host. The synthesis and assembly of well-defined cavities that bear endohedral or inwardly-directed functional groups are much less familiar.37 This is a consequence of the difficulty in performing reactions that result in functional groups—Lewis acids and bases—on concave molecular surfaces. Naturally-occurring receptors overcome this difficulty by arranging functional groups on a linear backbone then folding them around the target in a way that isolates the target from the medium and confronts it with the functional group. We have shown here that three 2,6-terphenyl acid ligands can present a UO22+ ion with carboxylate functions and surround it with aromatic panels. The hydrophobic environment and edge-to-face π-stacking of the capsule dramatically influences the solubility and kinetic stability of the resulting complex: the UO2+ ion can be extracted from aqueous solution even in the presence of considerable quantities of NaCl, and shows slow exchange of ligands on the 1-D NMR timescale. Given that some of the most versatile capsules use Pd or Pt ions in their assemblies,9 it may be prudent to consider capsule formation as a general means of precious metal sequestration. It is anticipated that these new chelating agents will selectively bind and sequester the uranyl ion from a variety of environments.Acknowledgements
We are grateful to the Skaggs Institute for Support. S.B. acknowledges the German Academic Exchange Association for a DAAD fellowship. O.B.B. is a Skaggs Institute Post-doctoral fellow. The M.G. Finn laboratory is thanked for the generous use of their ICP-AES spectrometer. Raj Chadha is gratefully acknowledged for his assistance with the crystallography.Notes and references
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 CrossRef CAS.
- J. M. C. A. Kerckhoffs, M. G. J. ten Cate, M. A. Mateos-Timoneda, F. W. B. van Leeuwen, B. Snellink-Ruël, A. L. Spek, H. Kooijman, M. Crego-Calama and D. N. Reinhoudt, J. Am. Chem. Soc., 2005, 127, 12697–12708 CrossRef CAS.
- L. R. MacGillivary and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS.
- K. Kobayashi, K. Ishii, S. Sakamoto, T. Shirasaka and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 10615–10624 CrossRef CAS.
- J. J. González, R. Ferdani, E. Albertini, J. M. Blasco, A. Arduini, A. Pochini, P. Prados and J. de Mendoza, Chem.–Eur. J., 2000, 6, 73–80 CrossRef CAS.
- J. Rebek, Jr., Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef; A. Scarso, L. Pellizzaro, O. De Lucchi, A. Linden and F. Fabris, Angew. Chem., Int. Ed., 2007, 46, 4972–4975 CrossRef CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS.
- (a) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusakawa and K. Birhada, Chem. Commun., 2001, 509–518 RSC; (b) Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS; (c) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- (a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS; (b) M. Ziegler, J. L. Brumaghim and K. N. Raymond, Angew. Chem., Int. Ed., 2000, 39, 4119–4121 CrossRef CAS.
- (a) F. J. Hurst, D. J. Crouse, Australian Patent 29312, 1971; (b) F. J. Hurst, D. J. Crouse, US Patent 3711591, 1973; (c) F. J. Hurst and D. J. Crouse, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 286 CrossRef CAS; (d) F. J. Hurst, D. J. Crouse, Canadian Patent 989623, 1976; (e) F. J. Hurst, D. J. Crouse and K. B. Brown, Ind. Eng. Chem. Process Des. Dev., 1972, 11, 122 CrossRef CAS; (f) U. M. J. Boin, The New Uraphos Process and the Mobile Pilot Plant, in Proceedings of a Technical Committee Meeting on Advances in Uranium Ore Processing and Recovery from Non-Conventional Resources, IAEA, Vienna, 1985, pp. 215–229 Search PubMed; (g) H. Gorecki, Extraction of Uranium(IV) from Wet-Process Phosphoric Acid with a Kerosene Solution of Nonylphenyl Phosphoric Acids, in Proceedings of a Technical Committee Meeting on Advances in Uranium Ore Processing and Recovery from Non-Conventional Resources, IAEA, Vienna, 1985, pp. 197–213 Search PubMed; (h) M. Sawicki, J.-M. Siaugue, C. Jacopin, C. Moulin, T. Bailly, R. Burgada, S. Meunier, P. Baret, J.-L. Pierre and F. Tara, Chem.–Eur. J., 2005, 11, 3689–3697 CrossRef CAS.
- J. R. Hart, Ethylenediaminetetraacetic Acid and Related Chelating Agents, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- A. Navaza, P. Charpin and D. Vigner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1842–1845 CrossRef.
- (a) P. G. Allen, J. J. Bucher, D. L. Clark, N. M. Edelstein, S. A. Ekberg, J. W. Gohdes, E. A. Hudson, N. Kaltsoyannis, W. W. Lukens, M. P. Neu, P. D. Palmer, T. Reich, D. K. Shuh, C. D. Tait and B. D. Zwick, Inorg. Chem., 1995, 34, 4797–4808 CrossRef CAS; (b) A. Coda, A. D. Giusta and V. Tazzoli, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1496–1500 CrossRef; (c) R. Graziani, G. Bombieri and E. Forsellini, J. Chem. Soc., Dalton Trans., 1972, 2059 RSC.
- T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 CrossRef CAS.
- B. M. Tadayoni, J. Huff and J. Rebek, Jr., J. Am. Chem. Soc., 1991, 113, 2247–2253 CrossRef CAS.
- K. D. Cramer and S. C. Zimmerman, J. Am. Chem. Soc., 1990, 112, 3680–3682 CrossRef CAS.
- J. A. Ippolito, R. S. Alexander and D. W. Christianson, J. Mol. Biol., 1990, 215, 457–471 CAS.
- P. H. Walton and K. N. Raymond, Inorg. Chim. Acta, 1995, 240, 593–601 CrossRef CAS.
- S. M. Butterfield and J. Rebek, Jr., J. Am. Chem. Soc., 2006, 128, 15366–15367 CrossRef CAS.
- B. W. Purse, S. M. Butterfield, P. Ballester, A. Shivanyuk and J. Rebek, Jr., J. Org. Chem., 2008, 73, 6480–6488 CrossRef CAS.
- M. D. Pluth, D. Fiedler, J. S. Mugridge, R. G. Bergman and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10438–10443 CrossRef CAS.
- A. R. Fersht, Enzyme Structure and Mechanism, WH Freeman, New York, 2nd edn, 1985, ch. 11, pp. 293–308 Search PubMed.
- C. J. F. Du, H. Hart and K. K. D. Ng, J. Org. Chem., 1986, 51, 3162–3165 CrossRef CAS.
- C. T. Chen and J. S. Siegel, J. Am. Chem. Soc., 1994, 116, 5959–5960 CrossRef CAS.
- Unfortunately, one of the 2,6-terphenyl carboxylic acid ligands is disordered preventing meaningful distance measurements within the crystal structure.
- (a) S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS; (b) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS.
- D. Lee and S. J. Lippard, Inorg. Chem., 2002, 41, 2704–2719 CrossRef CAS.
- Cambridge Structural Database, Version 5.27, November 2005, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK Search PubMed.
- Two iPr2EtN molecules are observed for each uranyl ion within this crystal structure. Because of the presence of the heavy uranium atom and the quality of the diffraction data, the absolute location of the hydroxyl and iPr2EtNH protons could not be determined. Consequently, we were unable to resolve whether the encapsulation complex has a charge of 1− or 2− (i.e. one deprotonated phenol and two counter cations or one counter cation and all phenols protonated).
- (a) J. J. Katz, L. R. Morss, G. T. Seaborg, The Chemistry of the Actinide Elements, Chapman and Hall, London, 2nd edn, 1986, pp. 1137–1140 Search PubMed; (b) K. W. Bagnall, Comprehensive Coordination Chemistry, Pergamon, Oxford, 1987, vol. 3, pp. 1187 Search PubMed.
- (a) G. R. Choppin and L. F. Rao, Radiochim. Acta, 1984, 37, 143–146 CAS; (b) P. F. Walch and D. E. Ellis, J. Chem. Phys., 1976, 65, 2387–2397 CrossRef CAS.
- (a) D. L. Clark, S. D. Conradson, R. J. Donohoe, D. W. Keogh, D. E. Morris, P. D. Palmer, R. D. Rogers and C. D. Tait, Inorg. Chem., 1999, 38, 1456–1466 CrossRef CAS; (b) J. A. Danis, M. R. Lin, B. L. Scott, B. W. Eichhorn and W. H. Runde, Inorg. Chem., 2001, 40, 3389–3394 CrossRef CAS; (c) D. M. Barnhart, C. J. Burns, N. N. Sauer and J. G. Watkin, Inorg. Chem., 1995, 34, 4079–4084 CrossRef CAS; (d) W. H. Zachariasen, Acta Crystallogr., 1948, 1, 281 CrossRef CAS; (e) W. H. Zachariasen, Acta Crystallogr., 1954, 7, 788 CrossRef CAS; W. H. Zachariasen, Acta Crystallogr., 1954, 7, 795 CrossRef CAS.
- M. J. Sarsfield and M. Helliwell, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS.
- While ligand exchange is a relatively slow processes in this system, the off diagonal signals observed in the ROESY experiments suggest that exchange is occurring in these reversible processes and the energy barrier to this exchange process is ΔG‡ = 17.1 kcal mol−1 (see ESI†).
- C. K. Vaughan, P. Harryson, A. M. Buckle and A. R. Fersht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 591–600 CrossRef.
- (a) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1083–1085 CrossRef CAS; (b) O. Seneque, M. N. Rager, M. Giorgi and O. Reinaud, J. Am. Chem. Soc., 2000, 122, 6183–6189 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of ligands 1–3 and their uranyl complexes with corresponding characterization, extraction studies, NMR titration and exchange experiments and X-ray diffraction data. CCDC reference numbers 760897 and 760898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00116c |
| ‡ Both authors contributed equally to this work. |
| § Crystal data [Et3NH]+[UO2(1)3]−·1.5CH2Cl2: C64.5H58Cl3NO8U, M = 1319.50, monoclinic, P21/n, a = 14.258(3), b = 18.699(4), c = 23.046(5) Å, α = 90.00°, β = 106.19(3)°, γ = 90.00°, V = 5901(2) Å3, Z = 4, ρc = 1.485 g mL−1, μ(Mo-Kα) = 2.941 mm−1, 2θmax = 50.00°, T = 153(2) K, 20725 reflections collected, R1 = 0.0610 for 6514 reflections, Rint = 0.0535 (668 parameters) with I > 2σ(I), and R1 = 0.0957, wR2 = 0.1765, GooF = 1.008 for all 9618 data, CCDC 760897. [iPr2EtNH]+·[UO2(33)]−·4H2O: C73H86N2O18U, M = 1517.47, monoclinic, Cc, a = 18.534(4), b = 19.636(4), c = 19.425(4) Å, α = 90.00°, β = 97.62(3)°, γ = 90.00°, V = 7007(2) Å3, Z = 4, ρc = 1.438 g mL−1, μ(Mo-Kα) = 2.387 mm−1, 2θmax = 50.00°, T = 296(2) K, 25087 reflections collected, R1 = 0.0549 for 9066 reflections, Rint = 0.0525, (864 parameters) with I > 2σ(I), and R1 = 0.0838, wR2 = 0.1428, GooF = 1.013 for all 12156 data, CCDC 760898. |
| This journal is © The Royal Society of Chemistry 2010 |