Discovery of an orexin receptor positive potentiator†
Jiyong
Lee‡
a,
M. Muralidhar
Reddy
a and
Thomas
Kodadek
*b
aDivision of Translational Research, Departments of Internal Medicine and Molecular Biology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390, USA
bDepartments of Chemistry & Cancer Biology, The Scripps Research Institute, Scripps Florida, 130 Scripps Way, Jupiter, FL 33458, USA. E-mail: Kodadek@scripps.edu; Tel: +1-561-228-2461
First published on 12th May 2010
Abstract
The orexin neurohormones control a variety of important physiological processes by signaling through two related G protein-coupled receptors, including appetite and feeding, wakefulness and energy homeostasis. Pharmacological manipulation of orexin signaling is an important goal. Here we describe the isolation of orexin receptor ligands from a library of microarray-displayed peptoids via a novel two-color, cell-based screen. Functional analysis of derivatives of these “hits” resulted in the development of moderate potency, low molecular weight receptor antagonists. Moreover, further optimization efforts resulted in the fortuitous discovery of a compound that positively potentiates the activity of the receptor. This compound is the first small molecule reported to up-regulate orexin signaling.
Introduction
The neurohormones orexin-A and orexin-B are produced in the hypothalamus in response to a drop in blood glucose levels. By signaling through two related G protein-coupled receptors, OXR1 and OXR2, the orexins regulate a number of important behaviors, including feeding,1,2 wakefulness,3,4 anxiety,5 reward seeking and addiction.6 Very recently, chronic stimulation of orexin signaling, mostly via OXR2, has been demonstrated to strongly resist high fat diet-induced obesity, hyperglycemia and hyperinsulinemia.7 This was shown to be due to increased leptin sensitization, increased energy expenditure and reduced food intake.Not surprisingly, there has been considerable interest in the development of compounds with which to manipulate orexin signaling. Non-peptide antagonists of OXRs have been reported by a number of groups (for a review, see ref. 8) and show promise in the treatment of insomnia.9 They may also be useful for the treatment of panic anxiety attacks5 and drug addiction,10 though no clinical trials for these indications have been carried out. On the other hand, activation of orexin signaling will be required to treat narcolepsy. The majority of human narcoleptics lack orexin-producing neurons, possibly due to an autoimmune attack on these cells,11 so in this case a full agonist will be required as a therapeutic lead.12 Based on the recent findings of Funato et al.7 an OXR2 receptor agonist might be useful in treating diet-induced obesity and diabetes. Since diabetics presumably produce normal physiological levels of orexin, a positive allosteric potentiator of receptor signaling might be interesting as well for this indication since it would hyperactivate orexin signaling in response to the native hormone.
To the best of our knowledge there have been no published reports of non-peptidic small molecules capable of stimulating orexin signaling. In this edge article, we describe the discovery of the first positive allosteric potentiator of the orexin receptors. This molecule resulted from the analysis of derivatives of modest potency orexin receptor antagonists that were isolated from a novel microarray-based screen.
Results and discussion
Array-based screening identifies a selective orexin receptor ligand
To identify orexin receptor ligands, we employed an assay that is an elaboration of previously reported cell-based receptor binding assays.13–15 As shown in Fig. 1, our plan was to display thousands of peptoids (N-substituted glycine oligomers)16 on a microarray platform, expose them to cells expressing the orexin receptor and identify peptoids that bind these cells with high selectivity. To encourage the isolation of selective ligands, we employed a two-color assay in which cells that do or do not express OXR1, but are otherwise identical, are exposed to the array and compounds that exhibit a strong preference for retention of the OXR1-expressing receptors are then identified.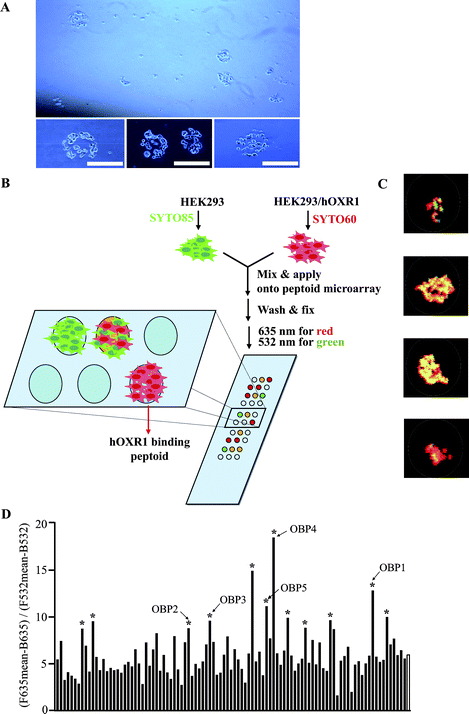 | ||
| Fig. 1 A microarray-based, two-color, cell-binding screen to isolate ligands of human OXR1. (A) Light microscopic images of cells binding to a peptoid microarray. Scale bar = 250 μm. (B) Schematic illustration of the screening procedure. (C) Representative superimposed images (red and green) of cells on a microarray after washing. (D) Ratiometric analysis of microarray images. The ratio of red to green fluorescence on 99 spots that displayed above background signal is shown. The unfilled bar at the far right of the graph represents the mean of the 99 spots. Bars marked with an asterisk represent peptoids that were subjected to sequence analysis by tandem mass spectrometry. Unambiguous sequences were obtained for peptoids marked OBP1–OBP5 (see ESI†). F635 and F532 are mean fluorescence emission intensities of spots with excitation wavelengths of 635 nm and 532 nm, respectively. B635 and B532 are background fluorescence emission intensities with 635 nm and 532 nm, respectively. | ||
To effect this strategy, we first examined if microarray-displayed peptoids17 are capable of binding to cells stably expressing human OXR1 (HEK293/hOXR1).18 The cells were incubated with a peptoid array (Fig. S1†) for one hour after pre-blocking the array with 3% BSA in DMEM to inhibit non-specific binding. The slide was then washed with PBS, and the binding of the cells to the array was examined under a microscope. As shown in Fig. 1A, circular patterns of cell monolayers were observed at certain points on the array and the diameters of the monolayers were similar to those of the printed peptoid spots (200–300 μm), suggesting binding of cells to peptoids spotted at these positions. This experiment suggested that the hybridization conditions were appropriate to assay for peptoid–receptor interactions.
HEK293 cells, which do not detectably express orexin receptors (as determined by Western blotting and quantitative RT-PCR; data not shown), were stained with SYTO 85 (green), and HEK293/hOXR1 cells were stained with SYTO 60 (red). After mixing the cells in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, they were applied to a peptoid microarray displaying 5760 different 9-mers and incubated for 1 h at 37 °C. After washing followed by fixation, the slide was scanned at 635 nm to visualize HEK293/hOXR1 binding and 532 nm to visualize HEK293 binding. The two images were then superimposed (Fig. 1B). We hypothesized that spots showing a high ratio of red over green fluorescence display peptoids that bind to OXR1 specifically. After measuring the level of fluorescence in each channel (532 nm and 635 nm, respectively) using a standard microarray scanner, we identified 99 spots displaying above background fluorescence in one or both channels (Fig. S2†). Some representative images of these spots are shown in Fig. 1C. Calculated ratios of the fluorescence intensities in the red and green channels are illustrated in Fig. 1D. The mean fluorescence ratio (635/532) of the 99 spots was 5.9. The fact that this ratio was not 1.0 was due to the optics of the scanner. We measured the fluorescence emissions ratio with 635 and 532 nm excitation wavelengths of a known 1:1 mixture of the differentially labeled cells and measured a ratio of 6.1. Thus, we concluded that the ratio of 5.9 reflects equal binding of the two cells on most of the features of the microarray. The twelve peptoid features displaying the highest red/green ratio above the mean were chosen as possible OXR1 ligands. Tandem MALDI mass spectrometry (using compound from the solution used to spot the microarray) provided unambiguous structures for five of these peptoids, which we named orexin receptor binding peptoids (OBPs; see Fig. S3†).
:1 ratio, they were applied to a peptoid microarray displaying 5760 different 9-mers and incubated for 1 h at 37 °C. After washing followed by fixation, the slide was scanned at 635 nm to visualize HEK293/hOXR1 binding and 532 nm to visualize HEK293 binding. The two images were then superimposed (Fig. 1B). We hypothesized that spots showing a high ratio of red over green fluorescence display peptoids that bind to OXR1 specifically. After measuring the level of fluorescence in each channel (532 nm and 635 nm, respectively) using a standard microarray scanner, we identified 99 spots displaying above background fluorescence in one or both channels (Fig. S2†). Some representative images of these spots are shown in Fig. 1C. Calculated ratios of the fluorescence intensities in the red and green channels are illustrated in Fig. 1D. The mean fluorescence ratio (635/532) of the 99 spots was 5.9. The fact that this ratio was not 1.0 was due to the optics of the scanner. We measured the fluorescence emissions ratio with 635 and 532 nm excitation wavelengths of a known 1:1 mixture of the differentially labeled cells and measured a ratio of 6.1. Thus, we concluded that the ratio of 5.9 reflects equal binding of the two cells on most of the features of the microarray. The twelve peptoid features displaying the highest red/green ratio above the mean were chosen as possible OXR1 ligands. Tandem MALDI mass spectrometry (using compound from the solution used to spot the microarray) provided unambiguous structures for five of these peptoids, which we named orexin receptor binding peptoids (OBPs; see Fig. S3†).
Characterization and improvement of the orexin receptor ligands
These five OBPs were re-synthesized, purified by HPLC and employed in functional assays. OXR1 receptor activation by orexin-A up-regulates adenyl cyclase, which subsequently increases cAMP production in OXR1-expressing cells.19 To quantify orexin-A induced cAMP elevation in HEK293/hOXR1 cells, we employed a reporter gene (3 × CRE-Luc) assay in which the promoter of the reporter gene contains a cAMP responsive element upstream of the firefly luciferase gene. As shown in Fig. S4A,† orexin-A stimulates cAMP elevation in a dose-dependent fashion (EC50 = 43 nM). This stimulation was blocked by the commercially available OXR1-selective antagonist SB408124 (Fig. S4B†) with an IC50 of 180 nM, which is similar to the reported value,20 validating that this assay properly measures orexin-triggered receptor activity.As shown in Fig. S5A,† none of the OBPs showed agonist activity, but some weak antagonist activity was observed (Fig. S5B†). For example, OBP1 showed an approximately 30% inhibition of orexin A-induced cAMP elevation at 300 μM. The activity of OBP2 was not quantified since it was cytotoxic to both HEK293 and HEK293/hOXR1 cell lines. The weak activity of OBP1 as an antagonist shows that the microarray-based binding assay is capable of registering even modest affinity receptor-binding molecules, probably because of avidity effects.
To further address the selectivity of OBP1 binding to OXR1, we utilized a different assay. OBP1 on Tentagel beads (TG-OBP1, Fig. S6†) were pre-incubated with 3% BSA in DMEM and then incubated with SYTO 60 stained-HEK293/hOXR1 cells or SYTO 60 stained-HEK293 cells, washed, and visualized by fluorescence microscopy. As shown in Fig. S7,† HEK293/hOXR1 cells showed binding to TG-OBP1 while HEK293 cells did not. This indicates that the peptoid does not bind stably to any other molecule on the surface of the HEK293 cells. We cannot rule out potential functional effects on other receptors without further study.
To determine if the peptoid recognizes the hormone-binding site of the receptor or some other surface of the extracellular region of the protein, we carried out a competition experiment. Whereas binding of HEK293/hOXR1 cells to TG-OBP1 was abolished by free OBP1, the addition of excess orexin A did not block binding of the cells to the immobilized peptoid (Fig. S7†). This suggests that OBP1 binds to a site distinct from that recognized by orexin itself and that it acts via an allosteric mechanism.
The low potency of OBP1 clearly must be improved for it to be a useful reagent. As a first step towards this goal, we sought to identify the minimal pharmacophore in the peptoid. Derivatives of OBP1 were synthesized in which each side chain (R) was replaced, in turn, with a methyl group (Fig. 2A and Fig. S8†).21 As shown in Fig. 2B, placement of the methyl group at the fifth and sixth positions of the peptoid (compounds OBP1-5 and OBP1-6) resulted in a complete loss of antagonist activity. Likewise, Tentagel beads displaying a peptoid in which both the Nmba and Npip residues of OBP1 were replaced with methyl groups did not show any binding to HEK293/hOXR1 cells (Fig. S7†). Finally, a truncated form of OBP1 (OBPt) containing only these two residues was found to exhibit antagonist activity equal to or even slightly better than the parent OBP1 peptoid (Fig. 2C). These results argue that the Nmba and Npip residues in the OBP1 hit comprise the minimal pharmacophore.
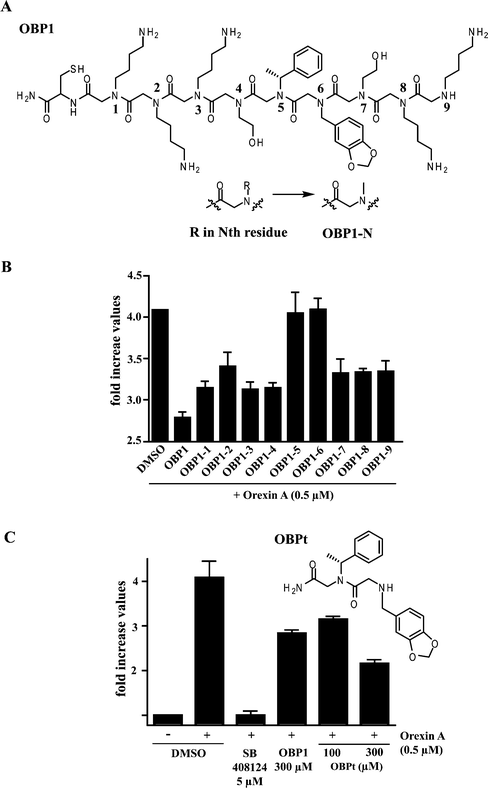 | ||
| Fig. 2 Pharmacophore identification via “sarcosine scanning”. (A) Each side chain (R) of the Nth residue was replaced, in turn, with a methyl group to afford OBP1-N (where N = 1–9), sarcosine containing peptoids. (B) Effects of sarcosine replacements on the antagonist activity of OBP1. The y axis shows the measured increase in cAMP concentrations in the cells relative to cells not treated with orexin (i.e., treatment with 0.5 μM orexin results in a four-fold stimulation of cAMP production). (C) Chemical structure of truncated OBP1 (OBPt) and its antagonist activity. Error bars represent the standard deviation of the mean of triplicate experiments. | ||
We noticed some structural similarity between OBPt and ACT-078573 (also known as Almorexant; see Fig. 3A), an orexin receptor antagonist developed by Actelion Pharmaceuticals.9 Based on this purported similarity, we hypothesized that appending a hydrophobic unit to the N-terminus of OBPt would place this group in approximately the same region of space as the CF3-substituted aryl ring in ACT-078573 and improve binding. The N-terminal secondary amine of OBPt was benzylated or benzoylated to afford OBPt-1 and OBPt-2, respectively (Fig. 3A). OBPt-1 was found to block orexin-A-induced cAMP elevation much more efficiently (IC50 = 20 μM; Fig. 3B) than OBPt. OBPt-2 showed a weaker, but still improved, activity (IC50 = 55 μM). Similar results were obtained by monitoring ERK phosphorylation, a known downstream mediator of OXR1 (Fig. 3C).19
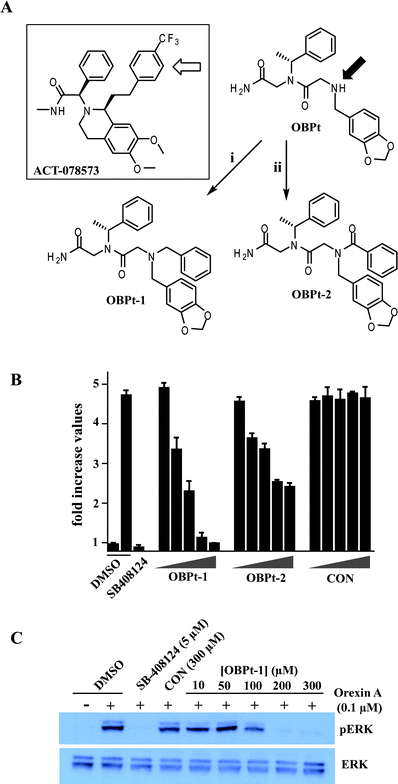 | ||
| Fig. 3 Effect of the introduction of hydrophobic groups at the N-terminus of OBPt on activity. (A) Comparison of the structures of ACT-098573, a potent antagonist of OXR1 and OXR2, and OBPt. The structures are oriented to illustrate the hypothesis that addition of an aryl substituent on the N-terminal nitrogen of the peptoid (marked with a filled arrow) would fill space that is occupied by the trifluoromethylphenyl-containing side chain in ACT-098573. (i) Benzaldehyde, BAP; (ii) benzoic acid, DIC, HOAt. (B) Antagonist activities of OBPt-1 and OBPt-2. Increasing concentrations (2, 20, 30, 50, and 75 μM) of peptoids were used. CON is an N-benzylated control peptoid (see Fig. S9†). Error bars represent the standard deviation of the mean from triplicate experiments. (C) Inhibition of orexin-mediated ERK phosphorylation by OBPt-1. A representative figure from three different experiments is shown. | ||
To identify even better compounds, a small library of additional derivatives was synthesized in which positions R1 through R4 (Fig. 4A) were varied. Detailed information about library construction and determination of the structure–activity relationships will be reported elsewhere. We found that OBPt-3 and OBPt-4 were more potent OXR1 antagonists than OBPt-1, with IC50 values of 4.5 μM and 15.1 μM, respectively (Fig. S10†). We then synthesized a derivative combining the two substitutions in OBPt-3 and OBPt-4 that distinguished them from OBPt-1 to afford OBPt-5. As shown in Fig. 4B, OBPt-5 showed increased potency (IC50 = 1.7 μM). The potency of the enantiomer OBPt-6 was almost identical (IC50 = 2.9 μM). Finally, we found that OBPt-5 also antagonized OXR2, which is 64% identical to OXR1 (Fig. 4C). OBPt-5 did not directly interfere with forskolin (adenyl cyclase activator)-induced cAMP production (Fig. S12†), which does not depend on orexin signaling. This argues that the antagonist effect of OBPt-5 was not due to some receptor-independent activity.
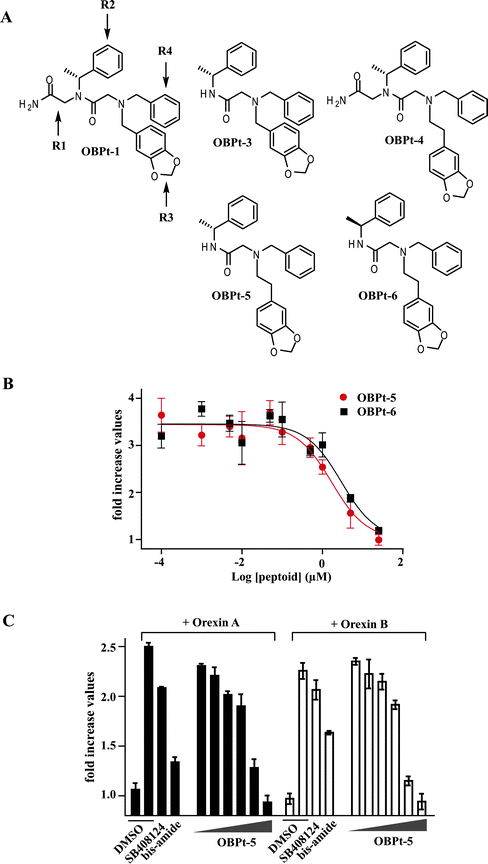 | ||
| Fig. 4 Attempted optimization of OBPt-1. (A) Chemical structures of some of the OBPt-1 derivatives examined. (B) Antagonist activities of OBPt-5 and OBPt-6. (C) Effect of OBPt-5 on orexin A- or orexin B-induced OXR2 activation of HEK293 cells expressing human OXR2. SB408124 is an OXR1 selective antagonist.20 Proline bis-amide is an OXR1/2 dual antagonist22 (Fig. S11†). | ||
Discovery of a positive potentiator or orexin receptor-mediated signaling
While testing various OBPt-1 derivatives we fortuitously identified three compounds (Fig. 5A) that did not antagonize the response of OXR1 to orexin A, but rather appeared to slightly enhance OXR1-mediated signaling in an orexin-responsive luciferase reporter gene assay (data not shown). This was somewhat surprising, given the very modest structural alterations in these compounds relative to OBPt-1. These original experiments employed concentrations of the orexin A peptide near the EC100. Therefore, to determine if these compounds are indeed OXR1 potentiators, we carried out experiments at 16 nM orexin A, which drives OXR1-mediated reporter gene expression in this assay at only about 20% of the maximum possible level (i.e., EC20 ≈ 16 nM). As shown in Fig. 5B, all of the compounds tested stimulated OXR1 activity under these hormone-limiting conditions. The most potent, OBPt-9, was chosen for further characterization. As shown in Fig. 5C, the maximal potentiation at this concentration of orexin A was about 2.5-fold, with an EC50 value for potentiation of approximately 120 nM. Note that a control peptoid (CON) did not show any activity. Importantly, OBPt-9 alone caused no activation of OXR1 (Fig. S13†).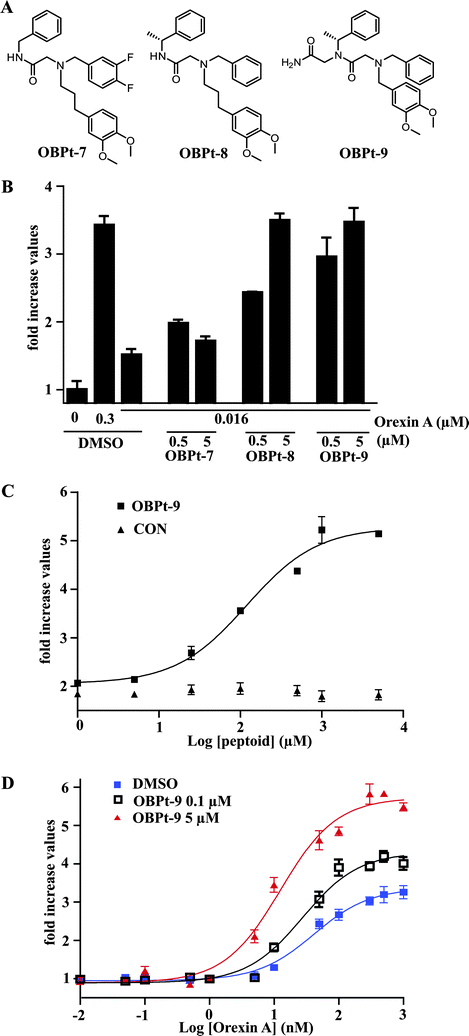 | ||
| Fig. 5 Discovery of a positive allosteric potentiator of the orexin receptors. (A) Chemical structures of tested compounds (OBPt-7, OBPt-8, and OBPt-9). (B) Effects of the compounds on the response (cAMP elevation) of OXR1-expressing cells to an EC20 concentration of orexin A. The level of cAMP elevation by 0.3 μM orexin A (the EC100 concentration) is also shown for comparison. (C) Concentration–response curves of OBPt-9 and CON on cAMP elevation of OXR1-expressing cells in the presence of an EC20 concentration of orexin A. (D) Concentration–response curves of orexin A on cAMP elevation of OXR1-expressing cells in the presence or absence of OBPt-9. | ||
We next determined the effect of OBPt-9 on the potency and efficacy of orexin A. Cells ware pre-incubated with OBPt-9 or DMSO (vehicle) and subsequently stimulated with increasing concentrations of orexin A. As shown in Fig. 5D, OBPt-9 induced a leftward and upward shift of the orexin A response curve. The EC50 value for orexin A in the presence of vehicle was 41 nM, whereas, the EC50 values were 27 and 12 nM in the presence of 0.1 μM and 5 μM of OBPt-9, respectively. Moreover, the maximal response to orexin A was about 2-fold higher in the presence of OBPt-9 (5 μM) than with vehicle alone.
Next, we examined if OBPt-9 can also potentiate the response of OXR2 to orexin A. After transient expression of OXR2, HEK293 cells also carrying a receptor-driven reporter gene were treated with OBPt-9 and then with increasing concentrations of orexin A. As depicted in Fig. S14,† OBPt-9 showed a similar potentiation pattern as was observed with the OXR1-containing cells. The orexin A EC50 was 60 nM in the presence of vehicle and 22 nM in the presence of OBPt-9. Moreover, the maximum level of reporter gene activation in the presence of OBPt-9 was almost twice that observed in the presence of vehicle. We also found that OBPt-9 did not affect ATP-dependent, endogenous P2 receptor-mediated ERK phosphorylation in HEK293 (Fig. S15†). This suggests the orexin receptor potentiation activity of OBPt-9 is not due to some non-specific cell surface receptor activation.
Conclusion
In summary, we have discovered new orexin receptor regulators using a microarray-based, two-color, cell-binding screen. An important feature of the screen is that it examines binding of prospective receptor ligands to differentially-labeled cells that do or do not display the target receptor but are otherwise identical. This allows the identification of compounds that display considerable selectivity for the target receptor, in contrast to typical screening methods that provide only information on potency, leaving selectivity issues to be worked out later. We also demonstrated a rapid and efficient route to pharmacophore identification and a preliminary optimization of the hit, which resulted in an antagonist with approximately 180-fold improved potency.Recently, we reported a similar two-color, cell-based screen carried out on Tentagel beads that allows up to several million peptoids to be screened simultaneously.15 While the microarray platform is limited to a few thousand compounds, it has the advantage of allowing facile and quantitative comparisons of the binding properties of all of the compounds on the array.23 Thus, while we demonstrated here the utility of microarrays for screening primary, naïve libraries, its most useful application may be in evaluating libraries of derivatives of primary hits in the search for improved ligands. Experiments along these lines are underway.
Importantly, we have discovered the first small-molecule allosteric potentiator of the orexin receptor. Allosteric potentiators bind to a site on the receptor distinct from that of the native ligand and accentuate the response of the receptor to that ligand, but cannot stimulate receptor function independently. It has been suggested that allosteric potentiators might have advantages over classical orthosteric agonists from the therapeutic point of view. For example, allosteric potentiators would not drive chronic receptor activation, but rather accentuate natural cycles of activation of the receptor.24,25 Animal experiments to test the utility of these compounds in vivo are underway.
Experimental
General remarks
All chemicals and solvents were purchased from commercial suppliers and used without further purification. Mass spectra were obtained with a Voyager-DE™ PRO (Applied Biosystems) for MALDI-TOF with an α-cyano-4-hydroxycinnamic acid matrix. MS/MS spectra were obtained on a 4700 Proteomics Analyzer (Applied Biosystems).Peptoid microarrays
Peptoid microarrays featuring 5760 different 9-mer peptoids were prepared as described before17 using the peptoid library shown in Fig. S1.†Cell culture
HEK293 cells were maintained in DMEM (Dulbecco's modified Eagle's medium, Invitrogen) supplemented with 110 mg L−1 sodium pyruvate, 2 mM L-glutamine and 10% (v/v) fetal calf serum at 37 °C in a 5% CO2 environment. For HEK293/hOXR1 cells, G418 (500 μg mL−1, Gibco) was included in the medium.A microarray-based, two-color, cell-binding screen and ratiometric image analysis
Cells were grown in culture plates to 95% confluency. The medium was replaced with a SYTO dye staining solution (5 μM in PBS, pH = 7.4) and incubated for 10 min at 37 °C. SYTO 85 was used for HEK293 and SYTO 60 was used for HEK293/hOXR1. Cells were washed with PBS three times and re-suspended with an incubation medium (3% BSA in DMEM). The two differentially stained cells were mixed at a 1:1 ratio in incubation media (10 mL), using 1 × 106 cells from each cell type. The cell suspension was added onto a microarray slide that had been washed with PBS and equilibrated with an incubation medium for 1 h. The Super PAP Pen (The Binding Sites, Inc.) was used to make a boundary along the edge of the microarray to hold cell suspension. After incubation for 1 h at 37 °C, the cell suspension was removed by suction and the microarray slide was placed on a Petri dish. PBS was added to cover the slide and the plate was gently shaken to wash off cells that bind nonspecifically to the microarray surface (30 s × 5 times). The microarray slide was then fixed (3% formaldehyde in PBS) for 10 min at room temperature and washed with PBS (10 s × 2 times). After brief washing with ddH2O, excess water was decanted and the microarray slide was dried with reduced exposure to light. The microarray slide was scanned with a ScanArray ExpressHT microarray scanner (PerkinElmer) using 532 nm (SYTO 85) and 635 nm (SYTO 60) lasers at 100% power and 300 PMT with a pixel size of 5. All the scanned images were analyzed by using the GENEPIX PRO 5.0 software (Axon Instruments, Union City, CA). Fluorescence intensities (F) of each spot at 635 nm or 532 nm after subtraction of mean local background intensity (B) were used for ratiometric analysis in which ratio = (F635 − B635)/(F532 − B532).
Peptoid synthesis
All peptoids were synthesized on Rink Amide AM resin (NovaBiochem) or Tentagel Macrobead NH2 resin (OBPt-3, OBPt-5, OBPt-6, OBPt-7, and OBPt-8. RAPP Polymere) using the sub-monomer approach by a microwave-assisted protocol26 and purified by preparative RP-HPLC (Waters) and confirmed with MALDI-TOF/MS. For the preparation of sarcosine derivatives (OBP1-1 to OBP1-9), methyl amine (2.0 M in THF) was used. For the preparation of CON, 2-methoxyethylamine was used. For OBPt-1, after addition of the second amine, the N-terminal secondary amine was benzylated under reductive alkylation conditions using benzaldehyde (10 eq.) and BAP (10 eq.). For OBPt-2, the N-terminal secondary amine was benzoylated with BzOH (3 eq.), DIC (3 eq.), and HOAt (3 eq.). All peptoids prepared on Rink Amide AM resin were cleaved from resins with cleavage cocktail (95% TFA, 2.5% water, 2.5% TIS) and purified by preparative RP-HPLC. See Table S1 for characterization of the synthesized peptoids.†Preparation of OBPt-3, OBPt-5, OBPt-6, OBPt-7, and OBPt-8
Tentagel Macrobead NH2 resin (RAPP Polymere) was reacted with 4-(bromomethyl)-3-nitrobenzoic acid (5 eq.) in the presence of DIC (5 eq.) and HOBt (5 eq.) for 3 h. After washing with DMF, the resin was reacted with (R)-, (S)-α-methylbenzylamine, or benzylamine in DMF (2 M) for 3 h. The resin was washed with DMF and then reacted with DIC (2 M) and bromoacetic acid (2 M) for 3 h. After washing with DMF, the resin was reacted with the corresponding second amine for 3 h to afford secondary amine. The resin was washed with DMF and then reacted with the corresponding benzaldehyde (10 eq.) and BAP (10 eq.) for reductive alkylation. After washing, peptoids were cleaved from the resin by exposing to UV (365 nm, 6 W) for 12 h in 2% v/v TFA/MeOH and purified by preparative RP-HPLC. See Table S1 for characterization of the synthesized peptoids.†Bead-binding assay
Peptoids were synthesized with Tentagel Macrobead NH2 resin (RAPP Polymere) as described previously15 and suspended in PBS. About 200 beads were dispensed into each well in 96-well plates. After incubation with 3% BSA in DMEM (100 μL) for 30 min at 37 °C, DMEM (100 μL) containing HEK293/hOXR1 or HEK293 cells (20000 cells) which were stained with SYTO 60 as described above were added. After 1 h incubation at 37 °C, the beads were washed with PBS (200 μL) five times before visualization under light/fluorescence microscopy. The Cy-5 channel was used to detect SYTO 60 emission.
Fluorescence measurement with plate reader
HEK293/hOXR1 and HEK293 cells were stained as described above with SYTO 60 and SYTO 85 and mixed in a 1:1 ratio in PBS. A total 5000 cells in PBS were added into a 96-well plate and the fluorescence emission at 670 nm and 575 nm were measured with excitation wavelengths of 635 nm and 532 nm, respectively. Three independent experiments were done to calculate the mean ratio of emissions at two wavelengths.
cAMP production assay
HEK293/hOXR1 cells were grown to ∼60% confluency in 48-well plates and transfected with the luciferase reporter plasmid, pGL3-3 × CRE-TATA (100 ng/well).27 24 h after transfection, the cells were serum-starved for 4 h and treated with the indicated concentrations of orexins. In the case of evaluating antagonists or allosteric potentiators, cells were pre-treated with indicated concentrations of peptoids for 20 min. 6 h after orexin addition, cells were lysed with passive lysis buffer (Promega) for 10 min at room temperature. To test agonist activity of peptoids, cells were treated with peptoids in the absence of orexin. The luciferase activity of the cell lysate was measured using the dual-luciferase assay kit (Promega) following the manufacture's protocol and normalized to Renilla luciferase activity from the co-transfected Renilla luciferase expression plasmid (pRLuc, 2 ng/well). Fold increase values of the normalized luciferase activities are presented in the figures. When HEK293 cells were used, the indicated concentration of forskolin was used instead of orexin. To examine the effect of peptoids on OXR2 expressing cells, HEK293 cells were transfected with phOXR2 (OriGene Technology Inc., 50 ng/well), pGL3-3 × CRE-TATA (50 ng/well), and pRLuc (1 ng/well) for 24 h before starvation.ERK phosphorylation assay
HEK293/hOXR1 cells were grown (∼75% confluency) in 6-well plates and serum-starved overnight prior to stimulation with orexin A for 12 min. For the antagonist evaluation, cells were treated with antagonists (SB408124 or peptoids) 20 min before the addition of orexin A. Cells were lysed with reporter lysis buffer (Promega) for 15 min at 4 °C. The collected lysates were mixed with 2 × SDS sample buffer and heated for 5 min at 95 °C. The samples were separated by SDS-PAGE and transferred to PDVF membranes (Immobilon, Millipore). The membranes were probed with anti-phospho ERK(1/2) or anti-ERK(1/2) primary antibodies (Santa Cruz) and subsequently developed with appropriate HRP-conjugated secondary antibody (BioRad) followed by chemiluminescence detection using SuperSignal® West Pico substrate (PIERCE). Quantifications of blot bands were performed using “Image J” software.Acknowledgements
This work was supported by a grant from the NIH (PO1-DK58398).Notes and references
- T. Sakurai, A. Amemiya, M. Ishii, I. Matsuzaki, R. M. Chemelli, H. Tanaka, S. C. Williams, J. A. Richardson, G. P. Kozlowski, S. Wilson, J. R. S. Arch, R. E. Buckingham, A. C. Haynes, S. A. Carr, R. S. Annan, D. E. McNulty, W.-S. Liu, J. A. Terrett, N. A. Elshourbagy, D. J. Bergsma and M. Yanagisawa, Cell, 1998, 92, 573–585 CrossRef CAS.
- L. de Lecea, T. S. Kilduff, C. Peyron, X. Gao, P. E. Foye, P. E. Danielson, C. Fukuhara, E. L. Battenberg, V. T. Gautvik, F. S. Bartlett, 2nd, W. N. Frankel, A. N. van den Pol, F. E. Bloom, K. M. Gautvik and J. G. Sutcliffe, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 322–327 CrossRef CAS.
- L. Lin, J. Faraco, R. Li, H. Kadotani, W. Rogers, X. Lin, X. Qiu, P. J. de Jong, S. Nishino and E. Mignot, Cell, 1999, 98, 365–376 CrossRef CAS.
- R. M. Chemelli, J. T. Willie, C. M. Sinton, J. K. Elmquist, T. Scammell, C. Lee, J. A. Richardson, S. C. Williams, Y. Xiong, Y. Kisanuki, T. E. Fitch, M. Nakazato, R. E. Hammer, C. B. Saper and M. Yanagisawa, Cell, 1999, 98, 437–451 CrossRef CAS.
- P. L. Johnson, W. Truitt, S. D. Fitz, P. E. Minick, A. Dietrich, S. Sanghani, L. Traskman-Bendz, A. W. Goddard, L. Brundin and A. Shekhar, Nat. Med., 2010, 16, 111–115 CrossRef CAS.
- P. J. Kenny, Trends Pharmacol. Sci., 2007, 28, 135–141 CrossRef CAS.
- H. Funato, A. L. Tsai, J. T. Willie, Y. Kisanuki, S. C. Williams, T. Sakurai and M. Yanagisawa, Cell Metab., 2009, 9, 64–76 CrossRef CAS.
- A. J. Roecker and P. J. Coleman, Curr. Top. Med. Chem., 2008, 8, 977–987 CrossRef CAS.
- C. Brisbare-Roch, J. Dingemanse, R. Koberstein, P. Hoever, H. Aissaoui, S. Flores, C. Mueller, O. Nayler, J. van Gerven, S. L. de Haas, P. Hess, C. Qiu, S. Buchmann, M. Scherz, T. Weller, W. Fischli, M. Clozel and F. Jenck, Nat. Med., 2007, 13, 150–155 CrossRef CAS.
- J. A. Hollander, Q. Lu, M. D. Cameron, T. M. Kamenecka and P. J. Kenny, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19480–19485 CrossRef CAS.
- J. Hallmayer, J. Faraco, L. Lin, S. Hesselson, J. Winkelmann, M. Kawashima, G. Mayer, G. Plazzi, S. Nevsimalova, P. Bourgin, S. S. Hong, Y. Honda, M. Honda, B. Hogl, W. T. Longstreth, Jr., J. Montplaisir, D. Kemlink, M. Einen, J. Chen, S. L. Musone, M. Akana, T. Miyagawa, J. Duan, A. Desautels, C. Erhardt, P. E. Hesla, F. Poli, B. Frauscher, J. H. Jeong, S. P. Lee, T. G. Ton, M. Kvale, L. Kolesar, M. Dobrovolna, G. T. Nepom, D. Salomon, H. E. Wichmann, G. A. Rouleau, C. Gieger, D. F. Levinson, P. V. Gejman, T. Meitinger, T. Young, P. Peppard, K. Tokunaga, P. Y. Kwok, N. Risch and E. Mignot, Nat. Genet., 2009, 11, 708–711 CrossRef.
- M. Mieda, J. T. Willie, J. Hara, C. M. Sinton, T. Sakurai and M. Yanagisawa, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4649–4654 CrossRef CAS.
- O. H. Aina, J. Marik, R. Liu, D. H. Lau and K. S. Lam, Mol. Cancer Ther., 2005, 4, 806–813 CrossRef CAS.
- O. H. Aina, T. C. Sroka, M. L. Chen and K. S. Lam, Biopolymers, 2002, 66, 184–199 CrossRef CAS.
- D. G. Udugamasooriya, S. P. Dineen, R. A. Brekken and T. Kodadek, J. Am. Chem. Soc., 2008, 130, 5744–5752 CrossRef CAS.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CAS.
- M. M. Reddy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12672–12677 CrossRef.
- D. Sikder and T. Kodadek, Genes Dev., 2007, 21, 2995–3005 CrossRef CAS.
- T. Holmqvist, L. Johansson, M. Ostman, S. Ammoun, K. E. Akerman and J. P. Kukkonen, J. Biol. Chem., 2005, 280, 6570–6579 CrossRef CAS.
- C. J. Langmead, J. C. Jerman, S. J. Brough, C. Scott, R. A. Porter and H. J. Herdon, Br. J. Pharmacol., 2004, 141, 340–346 CrossRef CAS.
- H.-S. Lim, C. T. Archer, Y.-C. Kim, T. Hutchens and T. Kodadek, Chem. Commun., 2008, 1064–1066 RSC.
- J. M. Bergman, A. J. Roecker, S. P. Mercer, R. A. Bednar, D. R. Reiss, R. W. Ransom, C. Meacham Harrell, D. J. Pettibone, W. Lemaire, K. L. Murphy, C. Li, T. Prueksaritanont, C. J. Winrow, J. J. Renger, K. S. Koblan, G. D. Hartman and P. J. Coleman, Bioorg. Med. Chem. Lett., 2008, 18, 1425–1430 CrossRef CAS.
- J. M. Astle, L. S. SImpson, Y. Huang, M. M. Reddy, R. Wilson, S. Connell, J. Wilson and T. Kodadek, Chem. Biol., 2010, 17, 38–45 CrossRef CAS.
- T. M. Bridges and C. W. Lindsley, ACS Chem. Biol., 2008, 3, 530–541 CrossRef CAS.
- P. J. Conn, A. Christopoulos and C. W. Lindsley, Nat. Rev. Drug Discovery, 2009, 8, 41–54 CrossRef CAS.
- G. M. Figliozzi, R. Goldsmith, S. C. Ng, S. C. Banville and R. N. Zuckermann, Methods Enzymol., 1996, 267, 437–447 CAS.
- P. G. Alluri, B. Liu, Y.P., X. Xiao and T. Kodadek, Mol. BioSyst., 2006, 2, 568–579 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. Supplementary tables and figures. See DOI: 10.1039/c0sc00197j |
| ‡ Present address: Department of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA. |
| This journal is © The Royal Society of Chemistry 2010 |