Task specific ionic liquids for the ionothermal synthesis of siliceous zeolites†
Paul S.
Wheatley
a,
Phoebe K.
Allan
a,
Simon J.
Teat
b,
Sharon E.
Ashbrook
a and
Russell E.
Morris
*a
aEaStChem School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: rem1@st-and.ac.uk; Fax: +44 1334 463808; Tel: +44 1334 463818
bAdvanced Light Source, Lawrence Berkeley National Lab, 1 Cyclotron Rd, mail stop 2-400, Berkeley, CA 94720, USA
First published on 25th June 2010
Abstract
The first genuine ionothermal synthesis of siliceous zeolites MFI and TON has been accomplished by utilising the ionic liquid 1-butyl-3-methyl imidazolium bromide/hydroxide as both solvent and structure directing agent.
Introduction
Zeolites are well established crystalline microporous materials possessing well-defined pore structures with ‘traditional’ applications in catalysis, ion exchange and gas separation, but also emerging applications1 in areas such as medicine.2,3 In order to maintain the momentum of future advances of zeolite chemistry, new synthetic routes to their crystallisation are in need of development. One such method developed recently is the ‘ionothermal’ route.At this time ionic liquids (ILs) are receiving enormous interest in many areas of chemistry.4 The majority of studies have reported on their potential ‘green’ chemistry attributes with particular interest as replacement for the solvent in homogeneous catalysis. The current definition of an IL encompasses compounds that are liquid at ambient temperatures and contain organic components.5 In the case of ionothermal synthesis, near room temperature ionic liquids (nRTILs) are defined as being liquid in the temperature range applicable to the synthetic conditions traditionally used in crystallisation of zeolites (e.g. at temperatures <200 °C).6,7 ILs have several notable properties that make them suitable media for the synthesis of inorganic materials,8,9 such as zeotypes. Depending on the choice of ionic liquid, they can be relatively polar in nature, therefore ensuring dissolution of the inorganic reagents.10,11 Many ionic liquids also possess good thermal stability enabling their use at elevated temperatures.
Previous studies from our laboratory reported the use of ILs for use as both solvent and as a structure directing agent (SDA) in the crystallisation of porous solids. This has produced many types of frameworks with varying compositions including zeolites12 and metal organic frameworks.13 The main advantage of ionothermal synthesis is the removal of competition between the solvent and the SDA for interaction with the growing crystal surface. It has been postulated that this may lead to improved templating of the growing zeolite framework14 and consequently to additional examples of ‘true’ templating.15 Ionothermal synthesis also takes place at ambient pressure because of the generally low vapour pressure of ionic liquids. In comparison, hydrothermal synthesis has both water and SDA in competition at the growing surface and generally occurs under high autogenous pressure.
To date, there are only a few accounts in the literature on the synthesis of silica-based materials using ionic liquids as templates in hydrothermal synthesis of zeolites16 and mesostructured silica,17 or as the solvent in the synthesis of silica aerogels.18 However, despite the success of ionothermal synthesis for the crystallisation of aluminophosphates there have been no examples of silica based zeolites prepared in this manner. The problems associated with the synthesis of siliceous zeolites from ILs can be attributed to the poor solubility of the silica starting materials. In this paper we show how these issues can be overcome by using an ionic liquid chosen because its chemistry mimics that which occurs in the traditional hydrothermal synthesis, allowing two siliceous zeolites to be prepared for the first time using an IL as both solvent and template.
A compelling advantage of ILs is that there is a wide range of chemistry available, leading to greater possibilities when it comes to tailoring the properties of the liquid towards a certain application. This has led to the concept of Task Specific Ionic Liquids (TSILs) where ionic liquids of specific properties are designed (or chosen) to match a desired application. Such designer solvents have been developed for use in a wide range of processes, from CO2 capture19 and other ‘green’ processes20 and catalysis,21 to the preparation of new carbon-based solids22 and the extraction of metal ions.23 This concept of designer properties is what truly makes ILs stand out as potentially extremely important solvents and reaction media. The challenge targeted in the work reported here is how to tailor the properties of ILs to make them more suitable for the preparation of silica zeolites.
As a starting point in this endeavour we looked at the types of IL that might mimic the conditions used in traditional hydrothermal24 or solvothermal25 zeolite synthesis. The first point for consideration is the nature of the IL cation, which is the intended template or structure directing agent in the ionothermal synthesis. Perhaps the most common type of cation used in ILs are imidazolium based, such as the 1-butyl-3-methylimidazolium cations shown in Fig. 1. While it has never been used itself in a zeolite synthesis, it is chemically similar to many other imidazolium-based cations (Fig. 1) that have been used in the synthesis of zeolites, especially by the group at ChevronTexaco in the USA.26,27
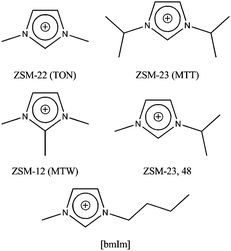 | ||
| Fig. 1 Examples of imidazolium-based cations that have been used successfully to prepare zeolitic materials. The structure of the ionic liquid cation (bmIm) used in this work is shown for comparison. | ||
Generally the reaction system in a zeolite synthesis either contains hydroxide28 or fluoride29 ions as mineralising agents to aid dissolution of the silicon-containing species. The simplest method used to accomplish this is the addition of a mineralising agent, such as hydroxide (e.g. NaOH) or fluoride (HF) to the solvent as has been done on many occasions. Unfortunately, despite many different attempts such approaches have not worked in ionothermal synthesis, the results either being non-crystalline solids or non-porous silica polymorphs.7
A more elegant approach would be to adapt the properties of the ionic liquid itself so that its properties were more suitable for the dissolution of silicon-containing reagents. The approach we demonstrate in this paper is that the partial exchange of the bromide anion of the IL for hydroxide leads to an IL that is chemically better suited for the dissolution of silica, while retaining the ability of the organic cation to template the resulting material. Having an IL combined with a hydroxide anion, together with an added fluoride mineraliser, aids the dissolution of silicate precursor and allows crystallisation to occur on a suitable timescale. Here we report the first ionothermal synthesis of siliceous zeolites Silicalite-1 (structure code MFI) and theta-1 (structure code TON) using 1-butyl-3-methylimidazolium hydroxide as the ionic liquid.
Experimental
Synthesis of 1-butyl-3-methylimidazolium hydroxide ([bmIm]OH)
The ionic liquid [bmIm]Br was synthesised according to previously published methods.13a The ion exchange of [bmIm]Br was carried out using an anion-exchange resin (Ambersep 900 OH form) in distilled water. Typically, the mass ratio of [bmIm]Br![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ambersep 900:water used was 1:1:4. The exchanges were carried out for at least 12 h, filtered, washed with distilled water and repeated again. The filtrate was then evaporated and the product dried under vacuum at 60 °C for 24 h to yield a brown viscous oil. NMR confirmed the presence of [bmIm] cation. 1H NMR (CDCl3): δ 0.95 (t, 3H, CH3, J = 7.3 Hz), 1.31–1.47 (m, 2H, CH2), 1.84–1.94 (m, 2H, CH2), 4.11 (s, 3H, NCH3), 4.33 (t, 2H, NCH2, J = 7.4 Hz), 7.45–7.46 (m, 1H, NC(H)C(H)N), 7.57–7.58 (m, 1H, NC(H)C(H)N, 10.26 (s, 1H, NC(H)N), but there is also a small amount of degradation of the IL. Reaction with silver nitrate confirmed the presence of residual bromide anions present after the ion exchange process. The amount of exchange is approximately 65%. The ionic liquid used in the reaction is therefore a ternary liquid of approximate formula [bmIm]OH0.65Br0.35
:Ambersep 900:water used was 1:1:4. The exchanges were carried out for at least 12 h, filtered, washed with distilled water and repeated again. The filtrate was then evaporated and the product dried under vacuum at 60 °C for 24 h to yield a brown viscous oil. NMR confirmed the presence of [bmIm] cation. 1H NMR (CDCl3): δ 0.95 (t, 3H, CH3, J = 7.3 Hz), 1.31–1.47 (m, 2H, CH2), 1.84–1.94 (m, 2H, CH2), 4.11 (s, 3H, NCH3), 4.33 (t, 2H, NCH2, J = 7.4 Hz), 7.45–7.46 (m, 1H, NC(H)C(H)N), 7.57–7.58 (m, 1H, NC(H)C(H)N, 10.26 (s, 1H, NC(H)N), but there is also a small amount of degradation of the IL. Reaction with silver nitrate confirmed the presence of residual bromide anions present after the ion exchange process. The amount of exchange is approximately 65%. The ionic liquid used in the reaction is therefore a ternary liquid of approximate formula [bmIm]OH0.65Br0.35
Synthesis of siliceous zeolites
Reactions were carried out in 23 mL Parr reaction vessels with the reactants mixed within the Teflon liners of these vessels.Typically [bmIm]OH0.65Br0.35 (4.3 g) is first added to the Teflon liner (23 mL capacity) along with TEOS (0.25 g, 1.20 mmol, Avocado, 98+%) and a small amount of distilled water (76.62 mg, 4.25 mmol) with stirring. After a period of time this mixture started to gel and HF (18.96 mg, 0.46 mmol, Aldrich, 48 wt% in H2O, 99.99+ %) was then added before the vessels were sealed and heated at 170 °C for 2 weeks. The products were recovered by filtration, washed well with distilled water, methanol, acetone and finally air dried.
The approximate molar starting composition was 20 IL:1 TEOS:4 H2O:0.38 HF, confirming that the IL is indeed the major solvent and that this is a true ionothermal preparation. However, there are significant amounts of molecular species present and some pressure is therefore evolved during the reaction, unlike many ionothermal synthesis where little pressure is released. The above preparation was repeated in a microwave assisted synthesis (Biotage Initiator) using half the stated quantities above (10 mL capacity reaction vessel). The vessel was heated to 170 °C for 1 h. This produced a maximum pressure of 6 bar during the reaction. The products were recovered in a similar manner to above.
Characterisation
Powder X-ray diffraction data were collected on a STOE STADIP diffractometer operating on monochromated Cu Kα1 radiation. Powder data were collected in Debye–Scherrer geometry using 0.5 mm quartz capillaries over a period of 15 h.The crystals recovered from all samples were too small for standard laboratory single-crystal X-ray diffractometers therefore diffraction data were collected on a Bruker AXS APEXII CCD area-detector diffractometer on station 11.3.1 of the Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA. The synchrotron beam was monochromated with a channel-cut Si(111) crystal providing a wavelength of λ = 0.77490 Å. Suitable crystals were mounted with paratone oil on a standard gonimeter head and cooled in a nitrogen gas cryostream flow using an Oxford Cryostreams low-temperature device operating at 150 K. Data reduction was carried out using Bruker SAINT package and synchrotron beam intensity decay was corrected using SADABS.30
Structures were solved by direct methods (SHELXS-97) and refined with full-matrix least-squares technique (SHELXL-97).31 All non-organic atoms were refined anisotropically and hydrogen atoms were geometrically placed. Details of data collections and structure refinement parameters are given in the Notes and references.‡ Full details of structure determinations are available in the ESI as CIF files.†
Solid-state NMR experiments were performed on Bruker Avance III 400 (13C and 29Si) or Bruker Avance III 600 (19F) spectrometers, equipped with widebore 9.4 or 14.1 T magnets respectively, at Larmor frequencies of 100.6 MHz for 13C, 79.5 MHz for 29Si and 564.7 MHz for 19F. For 13C/29Si experiments samples were packed in conventional 4-mm ZrO2 rotors and rotated at a rate of 10 kHz, whilst for 19F experiments the sample was packed into a 2.5-mm ZrO2 rotor and rotated at a rate of 30 kHz. Chemical shifts are recorded in ppm relative to TMS for 13C and 29Si and CFCl3 for 19F. For 13C, spectra were acquired using cross polarization, with a contact pulse (ramped for 1H) of 3 ms and 1H decoupling (SPINAL32 with ω1/2π = 100 kHz) applied throughout acquisition.
Thermogravimetric analysis was carried out using a TA Instruments SDT 2960 TGA/DTA analyser. Samples were heated in an aluminium crucible at a rate of 10 °C min−1 to a maximum temperature of 900 °C in a flowing atmosphere of oxygen (100 ml min−1). The organic content of the samples was determined using a Carlo Erba 1106 CHNS elemental analyser.
Results and discussion
The selection of a suitable IL was key to the successful crystallisation of siliceous zeolites. Previous attempts using ILs in their Br−/Cl− form have failed, even with the addition of fluoride or hydroxide ions as mineralising agents. In many of the recent successful attempts at the synthesis of siliceous zeolites, the organic additive has been utilised in its hydroxide form in hydrothermal synthesis. Therefore, exchanging the anion of the IL to that of hydroxide produces an organic additive that is similar. The resultant mixed bromide/hydroxide form, [bmIm]OH0.65Br0.35, is then used as both solvent and SDA in the synthesis of siliceous zeolites. Since the IL is in significant excess in the reaction we categorise this as the first true ionothermal synthesis of siliceous zeolites.As with many zeolite preparations, the results of the synthesis are relatively difficult to replicate, even given the design of the new IL for this use. Even when using the same, or extremely similar, conditions there are two different phases that can be prepared. For example, in one preparation phase pure Silicalite-1 (MFI framework topology) was crystallised, but when the same reaction conditions were repeated at a later date there were mixed products of Silicalite-1 and Theta-1 (framework topologies MFI and TON, respectively). In both cases the crystals were too small for data collection on a laboratory source so data were collected on a synchrotron source at the ALS, Berkeley. Full details of structure determination can be found in supplementary material.
[bmIm]-Silicalite-1
The structure determination of [bmIm]-Silicalite-1 (designated MFI framework by the International Zeolite Association) was completed in the usual orthorhombic space group of Pnma (Fig. 2). As with almost all previous samples of silicalite the crystal was heavily twinned along the crystallographic (110) direction. The framework contains twelve crystallographic non-equivalent silicon atoms and twenty-four oxygen atoms in the asymmetric unit. The Si–O distances and O–Si–O angles are all in the normal ranges expected for silica zeolites.32 Difference Fourier maps clearly indicate the presence of fluorine atoms (0.5 per unit cell, supported by NMR) as part of SiO4F within the pentasil units. The presence of the [bmIm] cation and ethanol (from hydrolysis of TEOS) within the pores of the structure was confirmed by 13C MAS NMR and found in the X-ray diffraction difference Fourier maps. The asymmetric unit contains 0.25 molecules of each of the organic molecules [bmIm]+ and ethanol. Chemical analysis and charge balancing considerations suggest that it is highly likely that there is additional disordered organic content that is not determined in the X-ray diffraction experiment. Silicalite-1 possesses a well-known pore structure composed of straight and sinusoidal 10-ring pores that intersect one another. There are many reports suggesting that there are three unique adsorption sites in Silicalite-1, the straight pores, the sinusoidal pores and their intersection points. It is at the intersections that the aromatic 5-ring structure of the imidazolium cations reside, at a relatively close approach to the occluded fluoride in the framework balancing the charge. This makes chemical sense as it is the fluoride that provides the negative charge to balance the positive charge of the [bmIm] cation. The butyl group of the imidazolium can be clearly seen protruding into the sinusoidal pore. The ethanol detected lies between imidazolium cations through the straight pore perpendicular to (010) direction.![The crystal structure of [bmIm]-Silicalite-1 viewed down the crystallographic c-axis. Single crystal X-ray diffraction reveals the location of the [bmIm] IL cation. Carbon, nitrogen, fluorine and silicon atoms are coloured black, blue, green and yellow, respectively. Oxygen atoms removed for clarity.](/image/article/2010/SC/c0sc00178c/c0sc00178c-f2.gif) | ||
| Fig. 2 The crystal structure of [bmIm]-Silicalite-1 viewed down the crystallographic c-axis. Single crystal X-ray diffraction reveals the location of the [bmIm] IL cation. Carbon, nitrogen, fluorine and silicon atoms are coloured black, blue, green and yellow, respectively. Oxygen atoms removed for clarity. | ||
13C solid-state MAS NMR (Fig. 3) of the material shows resonances in the expected regions for both the [bmIm] and the ethanol, confirming the nature of the species occluded in the zeolite pores suggested by the X-ray diffraction experiments. The presence of fluoride in the structure, again suggested by the X-ray diffraction experiment, is confirmed by 19F MAS NMR and 29Si MAS NMR shows the expected envelope of multiple resonances common for un-calcined silicalite-1 samples, with chemical shifts ranging from ∼−105 to −120 ppm.
![The 13C CP MAS NMR spectrum (9.4 T) of [bmIm]-Silicalite-1 confirming the presence of both ethanol and intact [bmIm] cations in the pores of the zeolite.](/image/article/2010/SC/c0sc00178c/c0sc00178c-f3.gif) | ||
| Fig. 3 The 13C CP MAS NMR spectrum (9.4 T) of [bmIm]-Silicalite-1 confirming the presence of both ethanol and intact [bmIm] cations in the pores of the zeolite. | ||
The chemical formula determined from the X-ray diffraction experiment is [Si48O96]F2(C8N2H15)(C2H7O). However, both elemental (experimental C 7.23%, H 1.31%, N 1.61%) and thermogravimetric analysis (weight loss 12.2%) suggest that there is additional organic content in the material that is not located in the X-ray diffraction experiment. On the basis of these experiments, together with the NMR, we suggest a better chemcial formula for the material is [Si48O96]F4(C8N2H15)2(C2H7O)2 which matches the elemental analysis extremely well (theoretical C 7.21%, H 1.33%, N 1.68%).
[bmIm]-Theta-1
As shown by both powder XRD and solid-state NMR, the [bmIm]-Silcialite-1 material described above can be prepared as a pure phase. However, sometimes another phase was formed as a mixture with [bmIm]-Silicalite-1. Single crystals of this phase could be separated from the solid, and X-ray diffraction showed the structure to be isostructural with the theta-1 zeolite framework (TON) determined in the usual orthorhombic space group of Cmc21 (Fig. 4). The asymmetric unit contains four crystallographically non-equivalent silicon atoms (two on special positions) and seven oxygens (two on special positions). Although there was residual electron density found within the channels, it could not be resolved into the [bmIm] cation or any other chemically sensible species. Subsequently, the SQUEEZE algorithm,33 which removes unmodelled electron density from the diffraction data, was applied. As this was a mixed phase, no other techniques were available to confirm the presence of intact [bmIm] within the pores of this material. However, it is notable that templating cations used to prepare TON frameworks previously are often disordered along the 1 dimensional channels present in this structure, making location by X-ray diffraction methods difficult or impossible.![The framework crystal structure of [bmIm]-theta-1. The templating cation location cannot be obtained from the XRD measurements. Silicon atoms are coloured yellow. Oxygen atoms removed for clarity.](/image/article/2010/SC/c0sc00178c/c0sc00178c-f4.gif) | ||
| Fig. 4 The framework crystal structure of [bmIm]-theta-1. The templating cation location cannot be obtained from the XRD measurements. Silicon atoms are coloured yellow. Oxygen atoms removed for clarity. | ||
Conclusions
In summary, for the first time siliceous zeolites, [bmIm]-silicalite-1 and [bmIm]-theta-1, have been synthesised using an ionothermal preparative method where an IL acts as both the solvent and structure directing agent/template. The success of the synthesis relied upon the preparation of a TSIL, [bmIm]OH0.65Br0.35, whose chemistry was designed to mimic that present in the traditional hydrothermal synthesis of zeolites. Such an example indicates that it is possible to alter the chemistry of ionic liquids so that they are suitable for the preparation of crystalline silica materials, something that had not been successful using this approach before. With careful consideration of the IL and its chemistry, this opens up a new route to the synthesis of siliceous zeolites.Acknowledgements
The authors thank the EPSRC for funding. R.E.M is a Wolfson Royal Society Merit Award Holder. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS.
- (a) H. B. Alam, Z. Chen, A. Jaskille, R. I. L. C. Querol, E. Koustova, R. Inocencio, R. Conran, A. Seufert, N. Ariaban, K. Toruno and P. Rhee, J. Trauma: Inj. Infect. Crit. Care, 2004, 56, 974–983 CrossRef CAS; (b) J. Galownia, J. Martin and M. E. Davis, Microporous Mesoporous Mater., 2006, 92, 61–63 CrossRef CAS.
- (a) H. A. Liu and K. J. Balkus, Chem. Mater., 2009, 21, 5032–5041 CrossRef CAS; (b) M. Mowbray, X. J. Tan, P. S. Wheatley, R. E. Morris and R. B. Weller, J. Invest. Dermatol., 2008, 128, 352–360 CrossRef CAS; (c) P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502–509 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS.
- K. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS.
- A. Taubert and Z. Li, Dalton Trans., 2007, 723–727 RSC.
- W. M. Reichert, J. D. Holbrey, K. B. Vigour, T. D. Morgan, G. A. Broker and R. D. Rogers, Chem. Commun., 2006, 4767–4779 RSC.
- P. Nockemann, B. Thijs, S. Pittois, J. Theon, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS.
- (a) R. E. Morris, Angew. Chem., Int. Ed., 2008, 47, 442–444 CrossRef CAS; (b) D. S. Wragg, P. J. Byrne, G. Giriat, B. Le Ouay, R. Gyepes, A. Harrison, A. G. Whittaker and R. E. Morris, J. Phys. Chem. C, 2009, 113, 20553–20558 CrossRef CAS; (c) E. A. Drylie, D. S. Wragg, E. R. Parnham, P. S. Wheatley, A. M. Z. Slawin, J. E. Warren and R. E. Morris, Angew. Chem., Int. Ed., 2007, 46, 7839–7843 CrossRef; (d) E. R. Parnham and R. E. Morris, J. Mater. Chem., 2006, 16, 3682–3684 RSC; (e) E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 4962–4966 CrossRef CAS; (f) L. Liu, Y. Li, H. B. Wei, M. Dong, J. G. Wang, A. M. Z. Slawin, J. P. Li, J. X. Dong and R. E. Morris, Angew. Chem., Int. Ed., 2009, 48, 2206–2209 CrossRef CAS.
- (a) Z. J. Lin, A. M. Z. Slawin and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 4880–4881 CrossRef CAS; (b) Z. J. Lin, D. S. Wragg and R. E. Morris, Chem. Commun., 2006, 2021–2023 RSC; (c) Z. J. Lin, D. S. Wragg, J. E. Warren and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 10334–10335 CrossRef CAS.
- R. E. Morris, Chem. Commun., 2009, 2990–2998 RSC.
- R. F. Lobo, S. I. Zones and M. E. Davis, J. Inclusion Phenom. Mol. Recognit. Chem., 1995, 21, 47–78 CAS.
- (a) X. Sun, J. King and J. L. Anthony, Chem. Eng. J., 2009, 147, 2–5 CrossRef CAS; (b) Y. Lorgouilloux, M. Dodin, J.-L. Paillaud, P. Caullet, L. Michelin, L. Josien, O. Ersen and N. Bats, J. Solid State Chem., 2009, 182, 622–629 CrossRef CAS.
- C. J. Adams, A. E. Bradley and K. R. Seddon, Aust. J. Chem., 2001, 54, 679–681 CrossRef CAS.
- (a) S. Dai, Y. H. Ju, H. J. Gao, J. S. Lin, S. J. Pennycook and C. E. Barnes, Chem. Commun., 2000, 243–244 RSC; (b) M. A. Klingshirn, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Mater. Chem., 2005, 15, 5174–5180 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- E. Lissner, W. F. de Souza, B. Ferrera and J. Dupont, ChemSusChem, 2009, 2, 962–964 CrossRef.
- J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- J. S. Lee, X. Q. Wang, H. M. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis and R. D. Rogers, Chem. Commun., 2001, 135–136 RSC.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–701 CrossRef CAS.
- R. E. Morris and S. J. Weigel, Chem. Soc. Rev., 1997, 26, 309–317 RSC.
- S. I. Zones and A. W. Burton, J. Mater. Chem., 2005, 15, 4215–4223 RSC.
- S. I. Zones, R. J. Darton, R. Morris and S. J. Hwang, J. Phys. Chem. B, 2005, 109, 652–661 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- (a) M. A. Camblor, L. A. Villaescusa and M. J. Diaz-Cabanas, Top. Catal., 1999, 9, 59–76 CrossRef CAS; (b) L. A. Villaescusa, P. S. Wheatley, I. Bull, P. Lightfoot and R. E. Morris, J. Am. Chem. Soc., 2001, 123, 8797–8805 CrossRef CAS.
- G. M. Sheldrick, Sadabs, Program for scaling and correction of area detector data, University of Göttingen, 1997 Search PubMed.
- SHELXS97 and SHELX97, Programs for Crystal Structure Analysis (Release 97-2). G. M. Sheldrick, Institüt für Anorganische Chemie der Universität, Tammanstrasse 4, D-3400 Göttingen, Germany, 1998 Search PubMed.
- D. S. Wragg, R. E. Morris and A. W. Burton, Chem. Mater., 2008, 20, 1561–1570 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnotes |
| † CCDC reference numbers 766491–766492. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00178c |
| ‡ Crystal data for [bmIm]-silicalite-1: [Si48O96]F2(C8N2H15)(C2H7O), M = 3108.62, λ = 0.77490 Å, orthorhombic, a = 19.959(5) Å, b = 19.890(5) Å, c = 13.407(3) Å, V = 5322.40(227) Å3, T = 150(2) K, space group Pnma, Z = 2, μ = 0.69 mm−1, 44702 reflections measured, 5948 independent reflections (Rint = 0.1814). The final R1 values were 0.0728 (I > 2σ(I)). The final wR(F2) values were 0.1838 (I > 2σ(I)). The final R1 values were 0.1410 (all data). The final wR(F2) values were 0.2115 (all data). The goodness of fit on F2 was 0.953. Crystal data for [bmIm]-theta-1: [Si8O16], M = 480.72, λ = 0.77490 Å, orthorhombic, a = 13.824(15) Å, b = 17.420(19) Å, c = 5.052(5) Å, V = 1216.7(39) Å3, T = 150(2) K, space group Cmc21, Z = 3, μ = 0.85 mm−1, 3798 reflections measured, 1244 independent reflections (Rint = 0.0996). The final R1 values were 0.104 (I > 2σ(I)). The final wR(F2) values were 0.2694 (I > 2σ(I)). The final R1 values were 0.1272 (all data). The final wR(F2) values were 0.2852 (all data). The goodness of fit on F2 was 1.105. |
| This journal is © The Royal Society of Chemistry 2010 |