Towards thermochromic and thermoresponsive near-infrared (NIR) luminescent molecular materials through the modulation of inter- and/or intramolecular Pt⋯Pt and π–π interactions
Kenneth Hoi-Yiu
Chan
ab,
Hoi-Shan
Chow
ab,
Keith Man-Chung
Wong
ab,
Margaret Ching-Lam
Yeung
ab and
Vivian Wing-Wah
Yam
*ab
aDepartment of Chemistry, The University of Hong Kong, Hong Kong
bInstitute of Molecular Functional Materials†, The University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: wwyam@hku.hk; Fax: +852 2857 1586; Tel: +852 2859 2153
First published on 25th June 2010
Abstract
A series of mononuclear and dinuclear alkynylplatinum(II) terpyridyl complexes has been found to show inter- and/or intramolecular aggregation in fluid solution state at low temperature or even at room temperature. Drastic colour changes and near infra-red (NIR) emission enhancement as well as broad and poorly resolved resonance signals in the 1H NMR spectra were observed. Such aggregation processes have been probed by 1H NMR, electronic absorption and emission spectroscopy.
Square-planar platinum(II) polypyridyl complexes represent a particularly interesting family of organometallic chromophoric complexes due to their intriguing spectroscopic and luminescence properties, as well as their propensity to exhibit metal-metal interactions.1–5 Platinum(II) terpyridyls, being an important class of this family, have been widely studied for their polymorphic behaviour and the luminescence colours associated with the extent of d8–d8 metal–metal interaction and π–π stacking between adjacent chromophoric units of the complexes.2–5
Since the first introduction of alkynyl ligands to the platinum(II) terpyridyl system by us in 2001,6 a number of reports on the studies of the spectroscopic and luminescence properties of this class of alkynylplatinum(II) terpyridyls have appeared.4,7 In particular, the self-assembly behaviour of these complexes has been demonstrated to be controlled by molecular design as well as changes in the microenvironment, such as solvent composition, counter-anions, polyelectrolytes and pH. One representative example of this class of versatile platinum(II) complexes is [Pt(terpyC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCCH]OTf (terpy = 2,2′:6′,2′′-terpyridine, OTf = trifluoromethanesulfonate).
CCCH]OTf (terpy = 2,2′:6′,2′′-terpyridine, OTf = trifluoromethanesulfonate).
Through appropriate design of dinuclear platinum(II) complexes of this class with flexible bridges on the terpyridyl ligand, interesting complexes that show a high propensity to form intramolecular Pt⋯Pt and π–π stacking interactions have been communicated previously.5 Aggregation and deaggregation of such dinuclear complexes could be controlled by modulation of the solution temperature. With our immense interest in the exploitation of the aggregation properties of platinum(II) alkynyl complexes and their associated electronic absorption and emission characteristics, a program has been initiated to understand the fundamental underlying principles that govern the supramolecular aggregation processes. Such studies would be anticipated to provide the basis for the better design of candidates that are capable of displaying obvious responses with changes in the environment. In this report, new excitatory findings regarding the temperature responsive aggregation behaviour of [Pt(terpy)CCCCH]OTf 1 and a new series of dinuclear complexes, [{(CH3)3SiCCCC}Pt{terpy-O(CH2CH2O)4-terpy}Pt{CCCCSi(CH3)3}](OTf)22, [(C18H37O-terpy)Pt{CCC6H4O(CH2CH2O)4C6H4CC}Pt(terpy-OC18H37)](OTf)23 (C18H37O-terpy = 4′-(n-octadecyloxy)-2,2′:6′,2′′-terpyridine), and [(tBu3-terpy)Pt{CCC6H4–O(CH2CH2O)4C6H4CC}Pt(terpy-tBu3)](OTf)24 (tBu3-terpy = 4,4′,4′′-tri(t-butyl)-2,2′:6′,2′′-terpyridine) (Scheme 1), investigated by temperature-dependent 1H NMR, electronic absorption and emission spectroscopy, will be described. All the complexes gave satisfactory elemental analyses and were characterized by 1H NMR spectroscopy and FAB mass spectrometry.‡
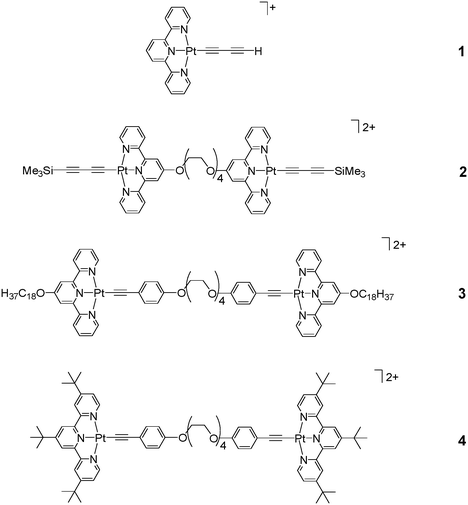 | ||
| Scheme 1 Structure of the mononuclear and dinuclear alkynylplatinum(II) terpyridyl complexes 1–4. | ||
At room temperature, the 1H NMR spectra of 1 and 4 in acetonitrile-d3 showed sharp signals with chemical shifts and splitting patterns consistent with their chemical formulation. On the contrary, 2 and 3 gave relatively broad and upfield-shifted signals in the aromatic region. Variable-temperature 1H NMR spectroscopy of 2 and 3 were performed and their spectra in acetonitrile-d3 are depicted in Fig. 1(a) and 1(b), respectively. The spectra recorded at elevated temperature were found to show sharp signals with the aromatic resonances shifted to the downfield region. In general, the spectra would show better resolved signals when the sample temperature was above 60 °C, whereas concentration and solvents seemed to have little effect on the 1H NMR spectra within the concentration range studied. The line broadening and upfield shifts of the aromatic signals in 2 and 3 at room temperature or below can be attributed to the self-assembly behaviour driven by the formation of Pt⋯Pt and π–π interactions in the solution aggregates, similar to that observed previously in a related system.5 Upon increasing the solution temperature, the Pt⋯Pt and π–π interactions would be destroyed and the observation of line sharpening as well as the downfield shifts of the 1H NMR aromatic resonances to their normal region could be attributed to the deaggregation process. On the contrary, bulky tert-butyl groups would exert large steric hindrance that prevents aggregation in 4 and this can account for the occurrence of sharp signals in the normal aromatic region in 4, which is different from its analogue 3 where aggregation would occur readily at room temperature.
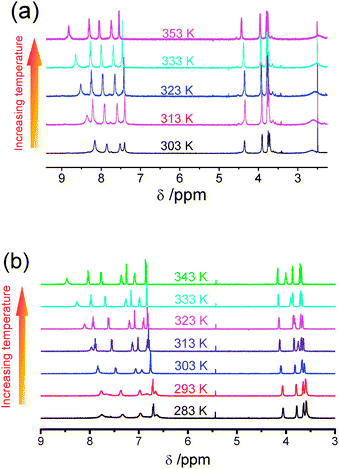 | ||
| Fig. 1 Variable-temperature 1H NMR spectra of (a) 2 and (b) 3 in acetonitrile-d3. | ||
Attempts to observe the induced aggregation of 1 in acetonitrile-d3 at low temperature were made by lowering the temperature in the 1H NMR spectroscopic study. The spectra became poorly resolved with an upfield shift of the aromatic resonances upon the gradual reduction of temperature to 213 K. Given the relatively high freezing point of acetonitrile-d3, acetone-d6 was used instead in this low-temperature 1H NMR study because of its lower freezing point and the spectra are shown in Fig. 2. The result is exciting and it brings to our attention that 1 could also form aggregates in solution by varying the temperature and the dimers or oligomers formed must be intermolecular in nature.
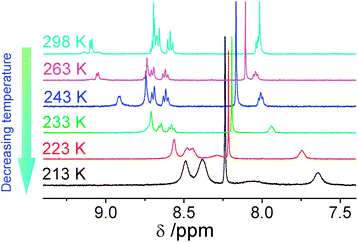 | ||
| Fig. 2 Variable-temperature 1H NMR spectra of 1 in acetone-d6. | ||
In order to further investigate the effect of temperature on the solution-state inter- and/or intramolecular aggregation properties of alkynylplatinum(II) terpyridyl complexes, variable-temperature studies on the electronic absorption and emission spectra of 1–4 were examined in this work.
In order to obtain a transparent medium at low temperature, butyronitrile was used as the solvent to replace acetonitrile in the electronic absorption and luminescence studies of 1. Dissolution of 1 in butyronitrile (3 × 10−4 M) gave a yellow solution at room temperature, while a dramatic colour change to greenish-blue occurred upon cooling the complex solution to form a butyronitrile glass at 77 K (Fig. 3). The electronic absorption spectra of the sample at 298 K and 77 K were measured. At room temperature, the low-energy absorption band was found at 412 nm whereas a new absorption band at 671 nm emerged when the sample was cooled to 77 K. The butyronitrile glass was warmed up until it melted completely and variable-temperature electronic absorption studies for the resulting sample solution were performed in the temperature range of 165 K to 298 K. It was found that 1 showed thermochromism, with the solution colour changing from blue to yellow upon increasing the temperature, and a drop in absorbance of the newly formed low-energy absorption band at 671 nm was found in the spectra with a concomitant growth of the MLCT band with well-defined isosbestic points (Fig. 4(a)).
 | ||
| Fig. 3 Photograph illustrating the colour change of 1 at room temperature (left) and low temperature (right) in butyronitrile upon cooling. | ||
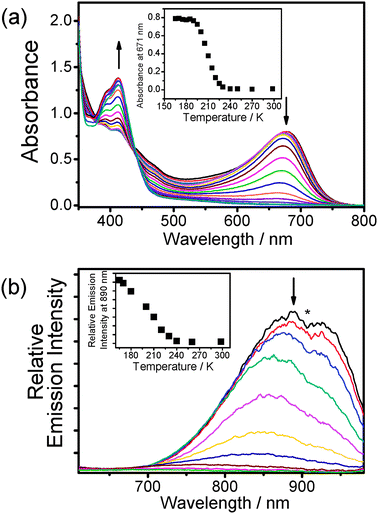 | ||
| Fig. 4 (a) UV-Vis spectra of 1 in butyronitrile with increasing temperature from 165 to 298 K. Inset: Plot of absorbance at 671 nm against temperature. (b) Corrected emission spectral changes of 1 in butyronitrile upon an increase in temperature from 165 to 298 K. Inset: Plot of emission intensity at 890 nm against temperature. The asterisk denotes instrumental artifacts. | ||
According to previous aggregation studies of 1 induced by the variation of solvent composition and by the electrostatic interaction with anionic polyelectrolytes and single stranded nucleic acids,3a–e together with its temperature-dependent 1H NMR study, the dramatic colour and spectral changes upon variation of temperature are ascribed to the self-assembly behaviour of the complex molecules. The low-energy absorption band formed at low temperature has been assigned to a metal–metal-to-ligand charge transfer (MMLCT) transition that arose from the close contacts of the platinum(II) metal centres, giving rise to intermolecular Pt⋯Pt and the ligand π–π interactions of the aromatic terpyridyl moieties in the glass state and low temperature solution state. As a result, the complex tends to form aggregates at low temperature provided that the sample solution is not at infinite dilution, and the degree of aggregation is sensitive to the change of temperature in the surroundings.
At 77 K, the butyronitrile glass of 1 showed a low-energy near infra-red (NIR) emission with λem at ca. 890 nm, and the corresponding variable-temperature emission studies in solution state were performed upon excitation at the isosbestic wavelength of 440 nm of the absorption spectral traces. Upon increasing the solution temperature from 165 K to 298 K, the NIR emission band at ca. 890 nm diminished in intensity and a slight blue shift in the emission maxima was also observed in the process (Fig. 4(b)). A complete “switch-off” of the NIR emission was found at ambient temperature, with the result in good agreement with that observed in the absorption studies. Similarly, the NIR emission at low temperature was tentatively assigned as 3MMLCT emission as a result of the intermolecular aggregation of 1.
Dilute solutions of 2, 3 and 4 in acetonitrile gave a yellow colour at 25 °C with similar UV-vis absorption patterns as that of 1 that include an intense absorption band at about 280–350 nm and a lower-energy absorption band at around 410–480 nm. With reference to spectroscopic studies of related platinum(II) systems,3–6 the higher-energy absorption bands in the UV region are assigned as intraligand (IL) [π → π*] transition of the terpyridine and alkynyl ligands, while the lower-energy absorption bands are assigned as metal-to-ligand charge transfer (MLCT) [dπ(Pt) → π*(terpy)] transitions, with some mixing of an alkynyl-to-terpyridine ligand-to-ligand charge transfer (LLCT) transition.
Variable-temperature photophysical studies of 2–4 were performed in acetonitrile. De-aggregation of these dinuclear platinum(II) complexes were predicted to occur at a much higher temperature than that of 1, as revealed by the temperature-dependent 1H NMR studies. In general, the effect of temperature on the colour and UV-vis absorption changes of 4 was negligible when compared to those of 2 and 3. At 233 K, 2 and 3 (3 × 10−4 M) showed an orange-red colour and a low-energy absorption band or shoulder appeared in the region of ca. 530–650 nm. A gradual change of solution colour to yellow occurred and the absorbance of the low-energy band would decrease upon increasing the temperature of the solutions. Eventually, the band would disappear when the temperature reached 343 K. According to our previous temperature-dependent studies on related dinuclear platinum(II) complexes5 and the similar findings observed in 1, these low-energy absorption bands beyond 540 nm observed at low temperature are attributed to MMLCT transitions. It is believed that the self-assembly of these dinuclear complexes is intramolecular in nature as a result of “sticking” together of the platinum(II) terpyridyl moieties in the molecule.
Owing to the good solubility of 2, a more concentrated solution sample (1.5 × 10−3 M) of 2 was prepared to investigate the nature of the solution aggregates. The concentrated solution sample of 2 in the temperature-dependent studies showed a more drastic colour change from blue-green to yellow upon an increase in temperature from 233 to 343 K. The temperature-dependent UV-vis spectral changes of 2 of different concentrations are depicted in Fig. 5. At 233 K, the MMLCT absorption band of 2 at higher concentration was found to red-shift to 624 nm, and interestingly, the absorbance of the MMLCT band dropped rapidly with a more-than-half reduction in absorbance for a temperature rise from 233 K to 258 K. The broad band at 624 nm then started to resolve into two bands at 546 and 644 nm, which could be observed for the concentrated sample at 258 K but diminished at a slower rate upon further temperature elevation. This is the first time that such differences in the diminishment of the absorbance of the MMLCT band in terms of a change in the rate and the band shape were observed. We suggest that the red-shifted λabs to 644 nm for the concentrated sample of 2 is due to the enhanced degree and extent of aggregation of 2 resulting from the co-existence of both the intra- and inter-molecular self-assembly processes in such conditions. As the low-energy 624 nm band was only observed in the concentrated sample solution of 2 but not in the dilute sample solution, together with the expectation that intermolecular aggregation should be more sensitive to temperature changes, the fast decay of the absorbance at 624 nm upon an increase in temperature from 233 to 258 K is believed to be a result of the rapid intermolecular disassembly process, as indicated in Fig. 5(b). Interestingly, the band at 546 nm coincided with the 542 nm MMLCT band in the low concentration regime, suggestive of its intramolecular origin. The change of band shape is an indication of the change of the nature of the aggregation, and the change of the rate of absorbance decay is also governed by the two aggregation pathways. When compared to 1 where only intermolecular aggregation could be possible, the similarity of the colour and the MMLCT band beyond 644 nm at low temperature may further confirm the intermolecular nature of the solution aggregates.
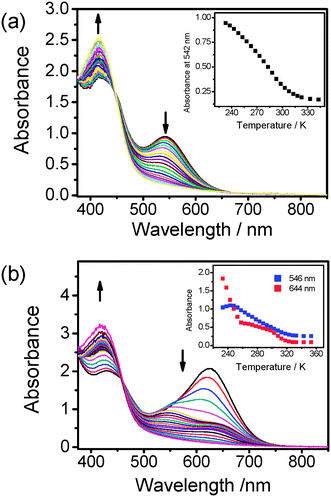 | ||
| Fig. 5 UV-Vis spectra of 2 in acetonitrile at different concentrations ((a) 3 × 10−4 M and (b) 1.5 × 10−3 M) with increasing temperature from 233 to 343 K. Inset: plot of absorbance of MMLCT band against temperature. | ||
Temperature-dependent UV-vis spectra of dilute sample solution of 3 in acetonitrile were also measured in the temperature range from 233 to 343 K, as shown in Fig. 5. Relative to 2, a less prominent absorption shoulder at about 574 nm emerges at low temperature, attributed to MMLCT transitions. The absorption shoulder showed a drop in absorbance with increasing temperature of the sample solution and approached zero upon heating to 343 K. The occurrence of a similar MMLCT absorption shoulder at 550 nm at low temperature was also observed in the related dinuclear complex with the CCC6H4-nBu-p substituents in the temperature-dependent UV-vis spectroscopic studies.5
Through the variation of temperature, the controllable intramolecular aggregation and de-aggregation processess of 2 under dilute conditions can be visualized as organometallic analogues of hairpin DNA with two sticky ends of platinum(II) terpyridyl units associated with the Pt⋯Pt and π–π interactions. With reference to the equilibrium studies of analogous hairpin DNA,8 the melting temperature (Tm), enthalpy change (ΔH°) and entropy change (ΔS°) of 2 for the de-aggregation process were calculated to be 276 K, +35 kJ mol−1 and +128 J mol−1 K−1, respectively, from the analysis of temperature-dependent UV-vis spectra in the dilute sample solution of 2 (Fig. 5(a)) by using a two-state van't Hoff model. It is interesting to note that similar thermodynamic parameters of melting temperature (275 K), enthalpy change (+32 kJ mol−1) and entropy change (+119 J mol−1 K−1) were obtained by monitoring the absorbance changes at 546 nm, which has been ascribed to the MMLCT absorption associated with the intramolecular aggregation process, in the corresponding temperature-dependent UV-vis spectra in the concentrated sample solution of 2 (Fig. 5(b)). The close resemblance of these two sets of data suggests that the intramolecular aggregation and deaggregation processess of this class of dinuclear complexes are concentration independent, in line with the corresponding variable-temperature 1H NMR spectroscopic studies. The melting temperature (299 K) and enthalpy change (+37 kJ mol−1) of the related dinuclear complex with the CCC6H4-nBu-p substituents5 were found to be slightly higher than those of 2, probably due to the presence of the sterically bulky SiMe3 groups in 2 which leads to a slightly weaker interaction between the two platinum(II) terpyridyl moieties. Attempts to probe the intermolecular aggregation behaviour of 2 by monitoring the absorbance of the 644 nm absorption band in the concentrated sample solution were unsuccessful because of the lack of a good fit of the experimental data to the two-state van't Hoff model, probably due to the complexity of the intermolcular aggregation process. Similarly, a melting temperature of 296 K, enthalpy change of +41 kJ mol−1 and entropy change of +140 J mol−1 K−1 were obtained for the de-aggregation process of 3 from the analysis of the data in Fig. 6. The comparable melting temperature and enthalpy change of 3 relative to 2 and the related dinuclear complex with the CCC6H4-nBu-p substituent indicate that the flexible bridging alkynyl ligand in 3 provides a similar function for the intramolecular aggregation as that of the bridging terpyridyl linkage. For the mononuclear complex 1, the melting temperature of 209 K is much lower than that of 2 and 3, suggesting that the presence of the flexible oligoether linkage in 2 and 3 would help to bring the two platinum(II) terpyridyl units into close proximity to form a more stable assembled structure.
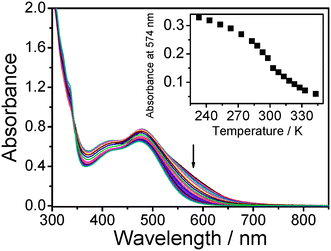 | ||
| Fig. 6 UV-Vis spectra of 3 in acetonitrile with increasing temperature from 233 to 343 K. Inset: Plot of absorbance at 574 nm against temperature. | ||
Upon excitation at the isosbestic wavelength observed in the variable-temperature UV-vis spectra, 2 (Fig. 7(a)) and 3 showed NIR emission bands with λem beyond 780 nm at low temperature. As the solution temperature was increased, the intensity of the NIR emission was found to gradually decrease with a slight blue shift of the band, in line with their corresponding UV-vis spectral changes and consistent with the variable-temperature studies of 1. The NIR emissions were attributed to originate from the 3MMLCT state as a result of self-association.
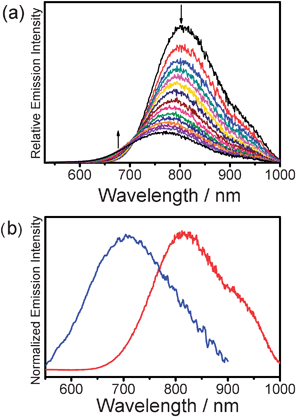 | ||
| Fig. 7 (a) Corrected emission spectral changes of 2 in acetonitrile (concentration = 1.5 × 10−3 M) upon an increase in temperature from 278 to 353 K. (b) Normalized emission spectra of 3 (red line) and 4 (blue line) in acetonitrile at room temperature. | ||
A dilute solution of 4 in deaerated acetonitrile gave a moderately intense emission at room temperature with λem of 712 nm upon excitation at λ > 400 nm (Fig. 7(b)). A blue shift of the emission energy was observed in 4 with three bulky tert-butyl groups on each of the terpyridyl units. The energy of the emission of 4 was not subject to change upon a variation of the solution temperature. As revealed in the 1H NMR spectroscopic study, sharp resonance signals with excellent resolution were observed at room temperature. This, together with the negative result from its variable-temperature emission study, suggested that the incorporation of bulky tert-butyl moieties in the terpyridyl ligand inhibits the self-assembly behaviour of 4 and hence the emission is originated from an excited state of 3MLCT character which has a higher energy than its analogue 3.
To conclude, a new class of dinuclear alkynylplatinum(II) terpyridyl complexes has been synthesized and characterized. Their aggregation properties, together with that of 1, have been probed by variable-temperature 1H NMR, electronic absorption and emission spectroscopic studies. Broad and poorly resolved 1H NMR signals were observed for 1–3 at low and/or room temperature. With the exception of 4, these complexes are found to be thermochromic and their solution colour was found to show drastic changes upon temperature variation in the range studied. Concentration was found to play an important role in governing the colour and the spectroscopic responses of 2. The spectral changes associated with the temperature alterations can be attributed to the inter- and/or intramolecular self-assembly behaviour of the complexes in the solution state. The driving force for this behaviour originates from the propensity of this class of compounds in forming Pt⋯Pt and π–π interactions. With judicious design, this may find useful applications as molecular temperature probes with tunable operation range.
V. W.-W. Y. acknowledges support from The University of Hong Kong under the Distinguished Research Achievement Award Scheme. This work has been supported by the University Grants Committee Areas of Excellence Scheme (AoE/P-03/08). K.H.-Y.C. and M. C.-L. Y. acknowledge the receipt of a University Postdoctoral Fellowship and a Postgraduate Studentship, respectively, both administered by The University of Hong Kong.
Notes and references
- (a) D. M. Roundhill, H. B. Gray and C. M. Che, Acc. Chem. Res., 1989, 22, 55–61 CrossRef CAS; (b) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529–1533 CrossRef CAS; (c) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS; (d) J. J. Novoa, G. AullVn, P. Alemany and S. Alvarez, J. Am. Chem. Soc., 1995, 117, 7169–7171 CrossRef CAS; (e) B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS.
- (a) K. W. Jennette, J. T. Gill, J. A. Sadownick and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 6159–6168 CrossRef CAS; (b) B. C. Tzeng, W. F. Fu, C. M. Che, H. Y. Chao, K. K. Cheung and S. M. Peng, J. Chem. Soc., Dalton Trans., 1999, 1017–1023 RSC; (c) H. K. Yip, C. M. Che, Z. Y. Zhou and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1992, 1369–1371 RSC; (d) H. K. Yip, L. K. Cheng, K. K. Cheung and C. M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933–2938 RSC; (e) W. Lu, M. C. W. Chan, N. Zhu, C. M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS; (f) J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1993, 32, 369–370 CrossRef CAS.
- (a) V. W.-W. Yam, K. M. C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS; (b) V. W.-W. Yam, K. H. Y. Chan, K. M. C. Wong and N. Zhu, Chem.–Eur. J., 2005, 11, 4535–4543 CrossRef; (c) C. Yu, K. M. C. Wong, K. H. Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791–794 CrossRef CAS; (d) C. Yu, K. H. Y. Chan, K. M. C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19652–19657 CrossRef CAS; (e) K. H. Y. Chan, J. W. Y. Lam, K. M. C. Wong, B. Z. Tang and V. W.-W. Yam, Chem.–Eur. J., 2009, 15, 2328–2334 CrossRef CAS; (f) C. Yu, K. H. Y. Chan, K. M. C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4577–4584 CrossRef CAS; (g) V. W.-W. Yam, Y. C. Hu, K. H. Y. Chan and C. Y. S. Chung, Chem. Commun., 2009, 6216–6218 RSC.
- M. X. Zhu, W. Lu, N. Zhu and C. M. Che, Chem.–Eur. J., 2008, 14, 9736–9746 CrossRef CAS.
- V. W.-W. Yam, K. H. Y. Chan, K. M. C. Wong and B. W. K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169–6173 CrossRef CAS.
- V. W.-W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 4476–4482 CrossRef CAS.
- (a) Q. Z. Yang, L. Z. Wu, Z. X. Wu, L. P. Zhang and C. H. Tung, Inorg. Chem., 2002, 41, 5653–5655 CrossRef CAS; (b) D. Zhang, L. Z. Wu, L. Zhou, X. Han, Q. Z. Yang, L. P. Zhang and C. H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS; (c) S. Chakraborty, T. J. Wadas, H. Hester, C. Flaschenreim, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6284–6293 CrossRef CAS; (d) F. Guo, W. Sun, Y. Liu and K. Schanze, Inorg. Chem., 2005, 44, 4055–4065 CrossRef CAS; (e) J. S. Field, R. J. Haines, D. R. McMillin, O. Q. Munro and G. C. Summerton, Inorg. Chim. Acta, 2005, 358, 4567–4570 CrossRef CAS; (f) R. Ziessel, S. Diring and P. Retailleauz, Dalton Trans., 2006, 3285–3290 RSC; (g) Y. Fan, Y. M. Zhu, F. R. Dai, L. Y. Zhang and Z. N. Chen, Dalton Trans., 2007, 3885–3892 RSC; (h) Z. Q. Ji, Y. J. Li and W. F. Sun, Inorg. Chem., 2008, 47, 7599–7607 CrossRef CAS.
- Y. Q. Shen, S. V. Kuznetsov and A. Ansari, J. Phys. Chem. B, 2001, 105, 12202 CrossRef CAS.
- (a) K. Sonogashira, S. Takahashi and N. Hagihara, Macromolecules, 1977, 10, 879 CrossRef CAS; (b) S. Takahashi, M. Kariya, T. Yakate, K. Sonogashira and N. Hagihara, Macromolecules, 1978, 11, 1063 CrossRef CAS; (c) V. W. W. Yam, C. H. Tao, L. Zhang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 453 CrossRef CAS.
Footnotes |
| † Areas of Excellence Scheme, University Grants Committee, Hong Kong. |
‡ Synthesis and Characterization. Complex 1 was synthesized and characterized according to the literature procedure previously reported by us.3a–e Complex 2 was synthesized by a modification of the Cu(I)-catalyzed reaction for alkynylplatinum(II) complexes.9 The dinuclear chloroplatinum(II) precursor, [ClPt{terpy-O(CH2CH2O)4-terpy}PtCl](OTf)2 (188.2 mg, 0.133 mmol) was dissolved in degassed N,N-dimethylformamide (10 mL) containing triethylamine (1 mL), and 1-(trimethylsilyl)-1,3-butadiyne (49.5 mg, 0.405 mmol) and a catalytic amount of copper(I) iodide were added. The reaction mixture was stirred for 48 h at room temperature, after which diethyl ether (100 mL) was added and stirred for 10 min. The precipitate that formed was isolated by filtration and washed with diethyl ether. Dichloromethane–acetone mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was used to dissolve the crude product and any insoluble residues were removed by filtration. Subsequent recrystallization of the complex was accomplished by diffusion of diethyl ether vapor into a red solution of the complex to give 2 as a dark brown solid. Yield: 123 mg (58%). 1H NMR (500 MHz, CD3CN, 333 K): δ (ppm) = 0.30 (s, SiMe3), 3.80–3.76 (m, 8H, OCH2), 3.94 (br, 4H, OCH2), 4.38 (br, 4H, OCH2), 7.46 (s, 4H, terpyridyl H), 7.69 (t, J = 5.6 Hz, 4H, terpyridyl H), 8.00 (d, J = 7.6 Hz, 4H, terpyridyl H), 8.28 (t, J = 7.6 Hz, 4H, terpyridyl H), 8.64 (d, J = 5.7 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1438 [M − OTf]+; elemental analysis, found: C 39.68, H 3.32, N 4.89; anal. calcd (%) for C54H54F6N6O11Pt2S2Si2: C 39.50, H 3.38, N 5.03. The procedure for the synthesis of 3 was similar to that described for the preparation of 2, except that the mononuclear chloroplatinum(II) precursor, [Pt(terpy-OC18H37)Cl]OTf (156 mg, 0.177 mmol) was used in place of [ClPt{terpy-O(CH2CH2O)4-terpy}PtCl](OTf)2 and HCCC6H4–O(CH2CH2O)4C6H4CCH (31.8 mg, 0.081 mmol) was used in place of 1-(trimethylsilyl)-1,3-butadiyne to give 3 as a dark brown solid. Yield: 104 mg (62%). 1H NMR (500 MHz, CD3CN, 333 K): δ (ppm) = 0.93 (t, J = 7.0 Hz, 6H, CH3), 1.49–1.33 (br, 60H, –CH2–), 3.75–3.68 (m, 8H, OCH2), 3.88 (m, 8H, OCH2), 4.18 (t, J = 4.5 Hz, 4H, OCH2), 6.86 (d, J = 8.6 Hz, 4H, –C6H4–), 6.98 (d, J = 8.6 Hz, 4H, –C6H4–), 7.16 (s, 4H, terpyridyl H), 7.26 (t, J = 6.8 Hz, 4H, terpyridyl H), 7.69 (d, J = 7.8 Hz, 4H, terpyridyl H), 7.98 (t, J = 7.8 Hz, 4H, terpyridyl H), 8.23 (d, J = 5.8 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1933 [M − OTf]+; elemental analysis, found: C 51.45, H 5.35, N 4.04; anal. calcd (%) for C92H18F6N6O13Pt2S2·CH2Cl2: C 51.49, H 5.58, N 3.87. The procedure for the synthesis of 4 was similar to that described for the preparation of 3, except that the mononuclear chloroplatinum(II) precursor, [Pt(terpy-tBu3)Cl]OTf (199.4 mg, 0.255 mmol) was used in place of [Pt(terpy-OC18H37)Cl]OTf. The reaction mixture was stirred for 48 h at room temperature; after which the solvent was removed under reduced pressure. The residue was subjected to column chromatography on silica gel using dichloromethane–acetone mixture (2:1 v/v) as the eluent. Subsequent recrystallization was accomplished by diffusion of diethyl ether vapor into a dichloromethane solution of the product to give 4 as an orange-red solid. Yield: 157 mg (71%). 1H NMR (400 MHz, CD3CN, 298 K): δ (ppm) = 1.44 (s, 36H, tBu), 1.49 (s, 18H, tBu), 3.67–3.61 (m, 8H, OCH2), 3.81 (t, J = 4.5 Hz, 4H, OCH2), 4.13 (t, J = 4.5 Hz, 4H, OCH2), 6.87 (d, J = 8.8 Hz, 4H, –C6H4–), 7.44 (d, J = 8.8 Hz, 4H, –C6H4–), 7.66 (dd, J = 1.9, 6.0 Hz, 4H, terpyridyl H), 8.12 (d, J = 1.9 Hz, 4H, terpyridyl H), 8.14 (s, 4H, terpyridyl H), 9.04 (d, J = 6.0 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1735 [M − OTf]+; elemental analysis, found: C 49.25, H 4.75, N 4.05; anal. calcd (%) for C80H94F6N6O11Pt2S2·CH2Cl2: C 49.41, H 4.91, N 4.27. :1 v/v) was used to dissolve the crude product and any insoluble residues were removed by filtration. Subsequent recrystallization of the complex was accomplished by diffusion of diethyl ether vapor into a red solution of the complex to give 2 as a dark brown solid. Yield: 123 mg (58%). 1H NMR (500 MHz, CD3CN, 333 K): δ (ppm) = 0.30 (s, SiMe3), 3.80–3.76 (m, 8H, OCH2), 3.94 (br, 4H, OCH2), 4.38 (br, 4H, OCH2), 7.46 (s, 4H, terpyridyl H), 7.69 (t, J = 5.6 Hz, 4H, terpyridyl H), 8.00 (d, J = 7.6 Hz, 4H, terpyridyl H), 8.28 (t, J = 7.6 Hz, 4H, terpyridyl H), 8.64 (d, J = 5.7 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1438 [M − OTf]+; elemental analysis, found: C 39.68, H 3.32, N 4.89; anal. calcd (%) for C54H54F6N6O11Pt2S2Si2: C 39.50, H 3.38, N 5.03. The procedure for the synthesis of 3 was similar to that described for the preparation of 2, except that the mononuclear chloroplatinum(II) precursor, [Pt(terpy-OC18H37)Cl]OTf (156 mg, 0.177 mmol) was used in place of [ClPt{terpy-O(CH2CH2O)4-terpy}PtCl](OTf)2 and HCCC6H4–O(CH2CH2O)4C6H4CCH (31.8 mg, 0.081 mmol) was used in place of 1-(trimethylsilyl)-1,3-butadiyne to give 3 as a dark brown solid. Yield: 104 mg (62%). 1H NMR (500 MHz, CD3CN, 333 K): δ (ppm) = 0.93 (t, J = 7.0 Hz, 6H, CH3), 1.49–1.33 (br, 60H, –CH2–), 3.75–3.68 (m, 8H, OCH2), 3.88 (m, 8H, OCH2), 4.18 (t, J = 4.5 Hz, 4H, OCH2), 6.86 (d, J = 8.6 Hz, 4H, –C6H4–), 6.98 (d, J = 8.6 Hz, 4H, –C6H4–), 7.16 (s, 4H, terpyridyl H), 7.26 (t, J = 6.8 Hz, 4H, terpyridyl H), 7.69 (d, J = 7.8 Hz, 4H, terpyridyl H), 7.98 (t, J = 7.8 Hz, 4H, terpyridyl H), 8.23 (d, J = 5.8 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1933 [M − OTf]+; elemental analysis, found: C 51.45, H 5.35, N 4.04; anal. calcd (%) for C92H18F6N6O13Pt2S2·CH2Cl2: C 51.49, H 5.58, N 3.87. The procedure for the synthesis of 4 was similar to that described for the preparation of 3, except that the mononuclear chloroplatinum(II) precursor, [Pt(terpy-tBu3)Cl]OTf (199.4 mg, 0.255 mmol) was used in place of [Pt(terpy-OC18H37)Cl]OTf. The reaction mixture was stirred for 48 h at room temperature; after which the solvent was removed under reduced pressure. The residue was subjected to column chromatography on silica gel using dichloromethane–acetone mixture (2:1 v/v) as the eluent. Subsequent recrystallization was accomplished by diffusion of diethyl ether vapor into a dichloromethane solution of the product to give 4 as an orange-red solid. Yield: 157 mg (71%). 1H NMR (400 MHz, CD3CN, 298 K): δ (ppm) = 1.44 (s, 36H, tBu), 1.49 (s, 18H, tBu), 3.67–3.61 (m, 8H, OCH2), 3.81 (t, J = 4.5 Hz, 4H, OCH2), 4.13 (t, J = 4.5 Hz, 4H, OCH2), 6.87 (d, J = 8.8 Hz, 4H, –C6H4–), 7.44 (d, J = 8.8 Hz, 4H, –C6H4–), 7.66 (dd, J = 1.9, 6.0 Hz, 4H, terpyridyl H), 8.12 (d, J = 1.9 Hz, 4H, terpyridyl H), 8.14 (s, 4H, terpyridyl H), 9.04 (d, J = 6.0 Hz, 4H, terpyridyl H); positive-ion FAB-MS: m/z 1735 [M − OTf]+; elemental analysis, found: C 49.25, H 4.75, N 4.05; anal. calcd (%) for C80H94F6N6O11Pt2S2·CH2Cl2: C 49.41, H 4.91, N 4.27. |
| This journal is © The Royal Society of Chemistry 2010 |