Catalytic asymmetric allylic alkylation employing heteroatom nucleophiles: a powerful method for C–X bond formation
Barry M.
Trost
*,
Ting
Zhang
and
Joshua D.
Sieber
Department of Chemistry, Stanford University, Stanford, CA 94305-5080, USA. E-mail: bmtrost@stanford.edu; Fax: +1 650-725-0002; Tel: +1 650-723-3385
First published on 18th June 2010
Abstract
Metal catalyzed asymmetric allylic alkylation (AAA) reactions have been an extensively studied and fruitful area of research in organic chemistry. The use of heteroatom-centered nucleophiles in this reaction is a powerful method for asymmetric C–X (X = heteroatom) bond formation. This perspective summarizes developments and applications of metal catalyzed AAA reactions employing heteroatom nucleophiles.
 Barry M. Trost | Born in Philadelphia, PA, in 1941, Professor Trost obtained his BA at the University of Pennsylvania (1962) and PhD at M.I.T. (1965). He directly moved to the University of Wisconsin where he became Professor in 1969 and Vilas Research Professor in 1982. He joined Stanford in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. Professor Trost has received a number of awards, including the ACS Award in Pure Chemistry (1977), the ACS Roger Adams Award (1995), the Presidential Green Chemistry Challenge Award (1998), the ACS Nobel Laureate Signature Award for Graduate Education in Chemistry (2002), and the ACS Cope Award (2004) in addition to election to the National Academy of Sciences (1990). He has published two books and over 850 scientific articles. |
 Ting Zhang | Ting Zhang obtained her BS degree in chemistry from Nankai University (China) in 2004. She then started her graduate studies with Professor Barry M. Trost at Stanford University, where her work focused on palladium-catalyzed asymmetric transformations in combination with other transition metal catalysis and their applications in complex molecule syntheses. After completion of her PhD degree in the summer of 2009, she joined the Medicinal Chemistry department at Merck & Co., Inc. in Rahway, New Jersey. |
 Joshua D. Sieber | Joshua D. Sieber obtained a BS in chemistry from Penn State University with honors in chemistry in 2003. He received a PhD from Boston College in 2008, where he worked under the direction of Professor James P. Morken and was an ACS Division of Organic Chemistry Fellow. He is currently an American Cancer Society Postdoctoral Fellow at Stanford University in the laboratory of Professor Barry M. Trost. |
Introduction
Methods for the installation of stereogenic carbon–heteroatom bonds in an enantioselective fashion are important for the preparation of chiral non-racemic amines, alcohols, thiols, and phosphines. Traditionally, formation of these bonds is usually effected by direct addition of a heteroatom nucleophile to a prochiral substrate such as an olefin (i.e. dihydroxylation, epoxidation, aziridination, etc.) or through addition of a nucleophile to a carbon–heteroatom double bond (i.e. carbonyl reduction, reductive amination, etc.). Over the past 30 years, transition-metal-catalyzed asymmetric allylic alkylation (AAA) reactions have been developed as powerful methods for the enantioselective formation of a variety of chemical bonds (i.e. C–C, C–O, C–N, C–S, C–P).1 While AAA reactions employing carbon nucleophiles have been extensively studied using a wide range of transition metal catalysts (Pd, Mo, Ir, Ru, Rh, Cu), AAA reactions employing heteroatom nucleophiles (i.e. N, O, S, P) are much less developed, especially with transition metal catalysts other than Pd. This perspective will highlight the development of AAA methodology employing heteroatom nucleophiles which has emerged as a powerful tool for asymmetric synthesis.Background
The successful use of heteroatoms as nucleophiles in AAA reactions is less reported than that of carbon nucleophiles. This fact partially results from the general challenges associated with the use of heteroatom nucleophiles in AAA reactions. First, heteroatoms can have significant affinities towards transition metals, resulting in the formation of stable chemical bonds which may inhibit catalytic turnover (i.e. halides, alkoxides), or outcompete the incoming substrate for complexation to the metal catalyst (i.e. phosphines). Second, heteroatoms are sometimes “harder” nucleophiles (i.e. alcohols, amines) compared to stabilized carbon nucleophiles, making the direct attack of the heteroatom on the “soft” metal π-allyl electrophile within the catalytic cycle more difficult (vide infra). Third, most heteroatom nucleophiles can also serve as good leaving groups (i.e. halides, imides, acetates), allowing for a reversible reaction that may result in loss of enantioselectivity. Due to the above factors, halides are generally considered unsuitable nucleophiles for AAA reactions, but for stabilized nitrogen, oxygen, and sulfur nucleophiles, experiments have been designed to overcome these barriers and achieve successful AAA reactions.Mechanism
The general catalytic cycle for the transition-metal-catalyzed AAA reaction is illustrated in Scheme 1.1 Initial binding of the metal to the olefin of the substrate, followed by ionization of the allylic leaving group leads to the intermediate π-allyl metal complexes 3 or 4, which may equilibrate through π–σ–π isomerization before nucleophilic addition. Importantly, the stereochemistry of the intermediate π-allyl complex formed after ionization is important for both diastereoselectivity and enantioselectivity in AAA reactions. Three possible configurations of the allyl ligand are possible: the syn,syn (3), the syn,anti (4), or the anti,anti (5). The π-allyl intermediate resulting from an E-olefin electrophile typically prefers the syn,syn configuration, whereas in a cyclic system, the π-allyl is locked in the anti,anti configuration. Lastly, trapping of these metal π-allyl complexes by the nucleophile then ensues to afford the product which is subsequently released from the catalyst.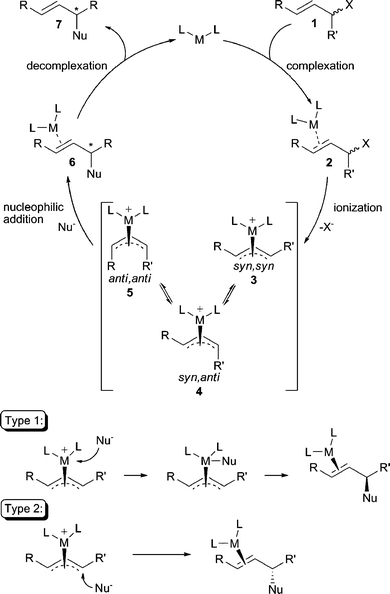 | ||
| Scheme 1 The standard AAA reaction mechanism. | ||
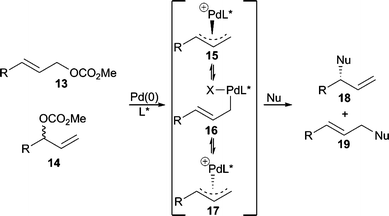
The nucleophilic substitution step in the AAA catalytic cycle can occur through two distinct mechanistic pathways that largely depend on the nature of the metal and nucleophile. That is, the nucleophile may first attack the metal center followed by subsequent reductive elimination to form the new bond (Type 1), or the nucleophile may attack the allylic carbon center directly, opposite from the face where the metal resides (Type 2). Nucleophilic attack via a Type 1 process results in retention2 of stereochemistry from the metal-allyl complex whereby reaction via a Type 2 process results in inversion3 of stereochemistry from the metal-allyl complex. In general, “hard” nucleophiles typically proceed through a Type 1 process, while “soft” nucleophiles proceed through the Type 2 process.
Enantiodiscrimination
Each step of the AAA catalytic cycle offers an opportunity for enantioselection. Therefore, several mechanisms for enantiodiscrimination are available to transition-metal-catalyzed AAA reactions. In AAA reactions with heteroatom nucleophiles, the following different mechanisms are proposed.One recent example of this mechanism can be found in the AAA reaction of methyl crotyl carbonate (8) with 4-methoxyphenol (Scheme 2).4 Here, enantioselectivity is significantly reduced with the addition of 30 mol% Bu4NCl. This finding was not simply a base effect, since employing a stronger base (K2CO3) led to no change in enantioselectivity relative to when no base was present. Furthermore, Bu4NCl is known to increase the rate of π–σ–π interconversion of the π-allyl Pd-complexes5 which leads to poor enantioselectivity in this case. Decreasing reaction concentration, which should presumably slow down Pd(0)–Pd(0) SN2 like exchange, also improved the reaction enantioselectivity. Furthermore, replacing the linear allylic carbonate (8) with the chiral racemic branched version led to low ee. All these findings suggest that fast π-allyl Pd-complex equilibration is unfavorable in this reaction, and therefore, reaction conditions that allow for fast trapping of the initially formed π-allyl Pd-complex lead to high enantioselectivity. Thus, the enantiodiscrimination step is believed to be the preferential ionization through enantioselective coordination of one of the prochiral olefin faces. Use of a less polar solvent (i.e. toluene), increased the enantioselectivity, an observation also consistent with this conclusion.
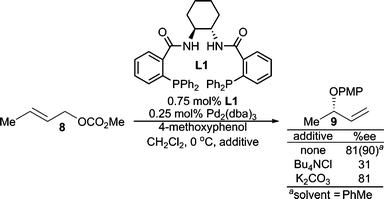 | ||
| Scheme 2 Enantioselection via enantiodiscrimination of the prochiral π-faces of 8. | ||
 | ||
| Scheme 3 Enantioselection via enantioselective ionization. | ||
 | ||
| Scheme 4 Enantioselection by discrimination of the π-allyl intermediates. | ||
The use of 1,3-diphenylallyl acetate (20, Scheme 5) in AAA reactions has become a standard test system for these processes.1a The ionization of 20 leads to the intermediate π-allyl complex 21, where the π-allyl ligand is now meso. When a chiral catalyst is employed, one terminus of the π-allyl ligand of 21 can be preferentially attacked by the nucleophile to afford one enantiomer of product. In this example, benzylamine serves as the nucleophile to afford an allylic amine product in excellent enantioselectivity.7
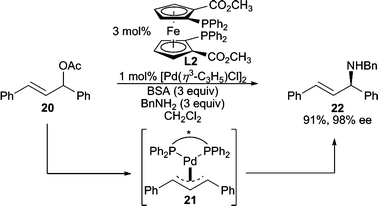 | ||
| Scheme 5 Deracemization through desymmetrization of an intermediate meso-π-allyl ligand. | ||
Amazingly, the chiral ligand resides on the opposite face of the π-allyl moiety relative to where the nucleophile attacks, yet it induces high enantiodiscrimination in these reactions. For Pd-catalyzed AAA reactions employing the Trost family8 of bidentate bis(phosphine) ligands (i.e.L1), this phenomenon has been explained empirically using a steric model.5b,9 However, recently, interactions between the nucleophile and the amide residues of the ligand backbone have been suggested to play a role.10 Additionally, chiral ligands containing a directing group for the nucleophile within the ligand backbone have been designed and implemented in AAA reactions.11 Conversely to bis(phosphines), when bidentate mixed donor ‘P,X’ ligands are employed, enantiodiscrimination has generally been rationalized12 by the “trans-effect.” That is, the allylic terminus of the π-allyl trans to the phosphino-moiety is more activated toward nucleophilic attack since the phosphine acts as a better acceptor ligand.
Nitrogen nucleophiles
Stereogenic C–N bonds are present in many bioactive molecules. Stereoselective construction of such bonds has been a constant challenge in organic synthesis. The development of AAA reactions with nitrogen-centered nucleophiles offers a significant opportunity for accessing such functionalities.Ammonia and amines
Ammonia has long been one of the most attractive nitrogen sources in allylic C–N bond forming reactions because chiral primary allylic amines are usually present in many target molecules. Applying ammonia as the reactant would be the most atom-economical16 approach in these cases. Surprisingly, very few examples employing ammonia directly in AAA reactions exist. The general challenges can be rationalized as: 1) ammonia can be an efficient ligand for many transition metal catalysts, and therefore, either prevents catalytic turnover or replaces the chiral ligand on the metal to generate achiral and sometimes non-reactive catalysts; 2) polyalkylation of ammonia is usually inevitable, and therefore, controlling the formation of monoalkylation is an issue; 3) ammonia is a significantly poorer nucleophile relative to substituted amines.Helmchen and co-workers17 have developed the use of diacylamine nucleophiles as NH3 surrogates in asymmetric Ir-catalyzed allylic amination. Hartwig and co-workers reported that NH3 in EtOH could be used as the nucleophile directly in an Ir-catalyzed AAA reaction with cinnamyl carbonate.18 In this system, while good branched regioselectivity is observed, the reaction does not stop at monoalkylation and affords only the dialkylated product in high dr and ee. More recently, Kobayshi and Nagano have reported a Pd-catalyzed AAA reaction using diphenyl allyl acetate and aqueous ammonia as the nitrogen source with BINAP as the chiral ligand (Scheme 6).19 Here, the monoalkylated product can be obtained in 71% yield and 87% ee. This result represents the first asymmetric Pd-catalyzed AAA reaction utilizing aqueous ammonia as a nitrogen source. The scope of this reaction has not been reported however.
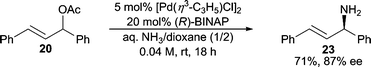 | ||
| Scheme 6 Use of NH3 in AAA reactions. | ||
Alkyl amines are reactive nucleophiles in AAA reactions, with somewhat slower reaction rates compared with stabilized carbon nucleophiles. In general, chemo-, regio- and stereoselectivities are the biggest challenges in AAA reactions employing alkyl amines as nucleophiles along with monoalkylation vs. bis(alkylation). For reactions with unsymmetrical allylic electrophiles, the regioselectivity in the AAA is important. Additionally, depending on the allylic substrate, the relative rate of nucleophilic attack vs. the rate of π–σ–π equilibration affects enantioselectivity. Thus, it is necessary to be at either extreme, i.e. rate of nucleophilic attack is much faster than the equilibration or vice versa.
Enantioselective allylic alkylation using alkyl amines was initially reported using symmetrical 1,3-diphenylallyl and 1,3-dialkylallyl carbonates by Hayashi and co-workers (Scheme 7).11 Notably, the attached hydroxy chain on the ligand is required for high enantioselectivity. Analysis of the X-ray crystal structure of the Pd-ligand complex revealed that the pendant hydroxy group is likely directing the nucleophile to attack a particular side of the allyl terminus. The same type of reaction can also be carried out using a PHOX type P,N-ligand as reported by Pfaltz, Helmchen, and co-workers.20
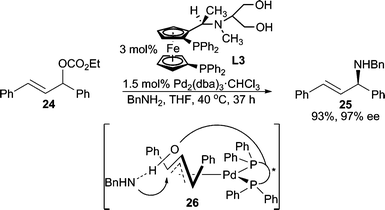 | ||
| Scheme 7 “Ligand”-directed nucleophilic attack. | ||
Use of α-aminoesters as nucleophiles using the modular Trost ligand L1 demonstrates the ligand control of regioselectivity when using 1,3-dialkylallyl carbonate electrophiles (Scheme 8).21 The chiral ligand can be used to overturn the inherent diastereoselectivity of the chiral non-racemic nucleophile with moderate diastereoselectivity in the mismatched case.
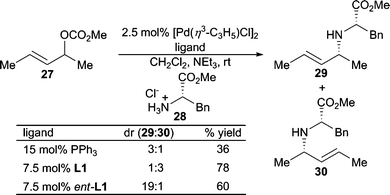 | ||
| Scheme 8 Ligand controlled diastereoselectivity. | ||
When using unsymmetrical allylic substrates, highly regio- and enantioselective AAA reactions are desired. Hayashi and Kishi demonstrated that both the E- and Z-isomers of crotyl acetate (31) gave good yields with high branched/linear selectivities in asymmetric allylic amination reactions (Scheme 9).22 The enantiomeric purity of the branched product is higher when using (E)-31. The reaction presumably proceeds through the Pd-π-allyl intermediates (32), which can undergo syn–anti isomerization (syn-32 and anti-32) and epimerization {(1R)-32 and (1S)-32} via π–σ–π interconversion. Hou, Dai and co-workers23 also developed a highly branched selective and enantioselective AAA reaction using BnNH2 as the nucleophile with a chiral ferrocene P,N-ligand.
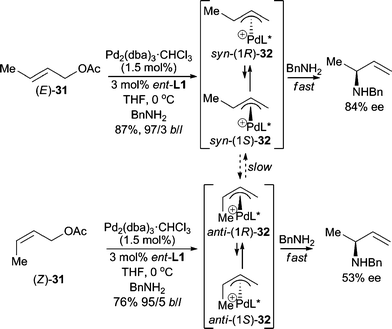 | ||
| Scheme 9 Allylic amination with BnNH2. | ||
In addition to Pd, other transition metals have also been investigated in allylic amination. Of note is the Ir-catalyzed allylic amination developed by Hartwig and Ohmura (Scheme 10).24 Good yields and excellent enantioselectivities are obtained with a variety of amine nucleophiles. High branched selectivity is observed, and in some cases, ≤4% of overalkylation is observed. Helmchen and co-workers have also developed asymmetric Ir-catalyzed intramolecular25 allylic amination processes and intermolecular allylic amination employing dienyl electrophiles.26 Additionally, planar chiral Ru-complexes have been demonstrated to effect AAA using nPr2NH as the nucleophile with moderate enantiocontrol (up to 74% ee).27
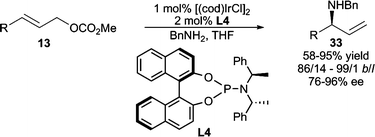 | ||
| Scheme 10 Ir-catalyzed AAA reaction with acyclic allylic carbonates. | ||
Aromatic amines
In contrast to alkyl amines, the use of aromatic amines as nucleophiles in AAA reactions is much less reported, presumably due to the lower nucleophilicity of an arylamine relative to an alkyl amine. One early example of a Pd-catalyzed asymmetric allylic amination using arylamines was reported by Hoberg and co-workers affording product in high enantiomeric excess in moderate yields.28 Interestingly, complementary regioselectivities were observed when using 4-methoxyaniline vs. aniline as the nucleophile. More recently, Hamada et al. have developed a Pd-catalyzed AAA reaction using arylamine nucleophiles with DIAPHOX, a novel diaminophosphine oxide preligand that is converted in situ from the P(V) form to the P(III) chiral ligand with BSA.29Asymmetric Ir-catalyzed allylic amination developed by the Hartwig group is also applicable to aromatic amines.30 Good yields and excellent regio- and stereoselectivities were obtained for a wide range of aromatic amine nucleophiles.30a Furthermore, in a recent report, the same group has developed an allylic amination procedure employing allylic alcohols as the electrophile (Scheme 11).31 This requires the addition of either a stoichiometric amount of Nb(OEt)5 or a catalytic amount of BPh3 to activate the allylic alcohol.
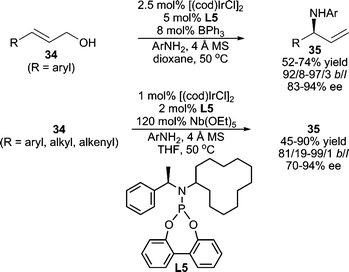 | ||
| Scheme 11 Ir-catalyzed AAA reactions employing allylic alcohols. | ||
Activated amides
Acetamides are typically poor nucleophiles for AAA reactions. The neutral amides are usually non-nucleophilic whereas the amide anions are strong bases and very hard nucleophiles that can sometimes deactivate the catalyst. One solution to this problem is to convert these “hard” nucleophiles into “softer” ones. This can be achieved, for example, by using potassium trifluoromethyl acetamide as the amide nucleophile with an Ir-catalyst.18 Similarly, N-alkoxy acetamides or hydroxylamines are effective nucleophiles in AAA reactions. Using Ir-catalysis with a Pybox derivative as the chiral ligand, allylic phosphonates undergo allylic amination with moderate regioselectivity and good enantioselectivity.32 Furthermore, a Pd-catalyzed asymmetric allylic amination utilizing an N-methoxy amide as nucleophile was employed for the total synthesis of agelastatin A.6 Cleavage of the N–O bond can later be achieved using SmI2 to give the formal amide addition compound.Isocyanates and carbodiimides
The use of isocyanates and carbodiimides for AAA has been developed for the preparation of diamines and aminoalcohols. For example, when vinyl aziridine 36 was treated with phenyl isocyanate, attack of the isocyanate by the aziridine leads to intermediate 37 after ionization, which subsequently cyclizes giving 38 (Scheme 12).33 This reaction also proceeds with vinyl epoxides and isocyanates or carbodiimides.34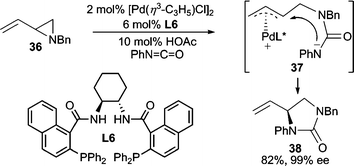 | ||
| Scheme 12 Pd-catalyzed AAA employing phenyl isocyanate. | ||
Azides
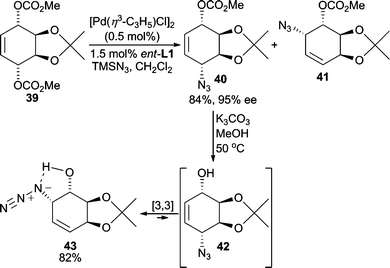
The azide functional group is a small and relatively strong nitrogen nucleophile for AAA reactions. The subsequent organic azide formed is typically easily converted to an amino group under mild reduction conditions. Furthermore, azides are relatively stable to a variety of reaction conditions. Thus, an azide group can be carried through multiple synthetic steps in organic synthesis and can thereby function as a “protected” amino group.Pd-catalyzed AAA reactions using TMSN3 as the nucleophile have been reported by Trost and Pulley.35 In the desymmetrization of meso-dicarbonate 39 (Scheme 13), the two regioisomers 40 and 41 were formed initially. However, lowering the catalyst loading minimized the formation of 41 allowing for isolation of 40 in 84% yield. This concentration effect suggests that the Pd-catalyst may be catalyzing the isomerization of 40 to 41, or that a [3,3]-sigmatropic rearrangement process that is common among allylic azides occurs to some extent.35 Because of this [3,3]-rearrangement of allylic azides, preparation of regioisomerically pure allylic azides is sometimes difficult. In this system, either allylic azide is accessible since saponification of 40 at 50 °C leads to a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomeric allylic azides 43 and 42, respectively. Hydroxyazide 43 is isolated in 82% yield and is presumably favored due to the hydrogen bond shown. Thus, this method renders the options of forming 1,4- or 1,2- substituted products selectively.36
:1 mixture of regioisomeric allylic azides 43 and 42, respectively. Hydroxyazide 43 is isolated in 82% yield and is presumably favored due to the hydrogen bond shown. Thus, this method renders the options of forming 1,4- or 1,2- substituted products selectively.36
 | ||
| Scheme 13 Azide nucleophiles in AAA. | ||
Sulfonamides
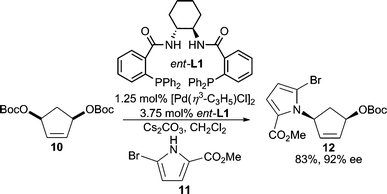
Both alkyl and free sulfonamides are good nucleophiles for AAA reactions. The highly electron-withdrawing nature of the sulfonyl group results in “softening” of the nitrogen center. For example, p-toluenesulfonamide was shown to be effective in an AAA reaction of a cyclic allylic carbonate giving the product in good yield and enantioselectivity;37 while an alkyl substituted secondary nosyl sulfonamide38 was used in the desymmetrization of a 5-membered cyclic meso-carbonate. Sulfonamide nucleophiles have been widely used in AAA.11a,12,20,39 Additionally, in an interesting intramolecular Pd-catalyzed AAA reaction (Scheme 14), poor reactivity was initially observed when using the standard Trost ligand (ent-L1).37 This result led the authors to design a new ligand system (L7) based on the working model for selectivity in AAA reactions with the Trost family of ligands.9 Gratifyingly, this mixed P,N-ligand system allowed for the preparation of the desired product in high yield and enantioselectivity. This protocol allowed for the asymmetric synthesis of (−)-anatoxin-a.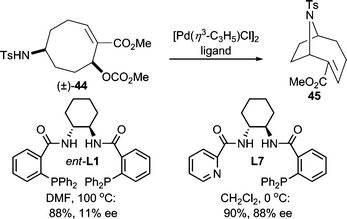 | ||
| Scheme 14 Modified Trost-ligand for improved enantioselection in the intramolecular Pd-catalyzed AAA of rac-44. | ||
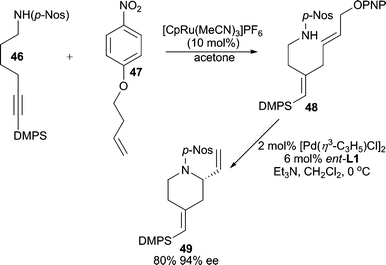
Utilizing a tandem Ru-enyne cross-coupling/intramolecular Pd-AAA process, a variety of nitrogen and oxygen containing heterocycles can be synthesized (Scheme 15).40 A p-nitrobenzenesulfonyl amide must be used as the nucleophile for the AAA reaction because the p-Nos moiety is tolerant of the Ru-catalyzed coupling reaction. Furthermore, a p-nitrophenoxy (PNPO) activated allylic alcohol is the optimum leaving group for the electrophilic coupling partner in the AAA.
 | ||
| Scheme 15 Tandem Ru-catalyzed enyne cross-coupling/intramolecular AAA. | ||
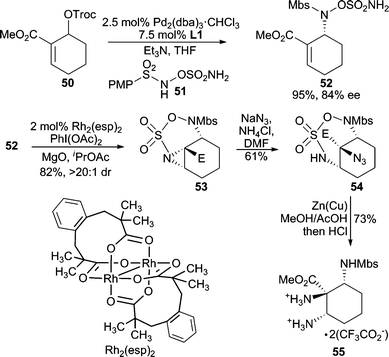
Very recently, hydroxylamine-O-sulfamates (i.e.51, Scheme 16) have been applied as nucleophiles in tandem Pd-AAA/Rh-aziridination reactions.41 This nucleophile class participates in chemoselective AAA reactions, where the hydroxylamine functionality preferentially acts as the nucleophile over the sulfamate moiety producing 52. Subsequently, the sulfamate group can be employed in intramolecular Rh-catalyzed aziridination reactions to afford, for example, 53 in excellent diastereocontrol. Opening of the aziridine with NaN3 preferentially occurs at the more substituted carbon atom of the aziridine, and subsequent removal of the sulfonyl group with Zn(Cu) couple allows for efficient access to unnatural triaminoester 55 in enantioenriched form.
 | ||
| Scheme 16 Tandem Pd-catalyzed AAA/Rh-catalyzed aziridination. | ||
Imides
Imides have been extensively employed as nucleophiles for AAA reactions due to their “soft” character which results from delocalization of electron density on nitrogen into the attached carbonyl or sulfonyl groups of the imide. Among all imide nucleophiles, phthalimide is the most commonly used nitrogen nucleophile in AAA reactions because of its excellent reactivity, and because the phthaloyl group on the resulting allylic amine product serves as a convenient protecting group for the amino moiety. Cyclic allylic acetates,42 vinyl epoxides,43 and divinyl carbonate44 have all been effectively employed in Pd-catalyzed AAA reactions in high enantioselectivities using the Trost family of chiral ligands.A unique cyclic allylic lactone electrophile (56, Scheme 17) has been used in a variety of AAA reactions for C–C and C–O bond forming reactions; however, its use in analogous C–N bond forming reactions with imides gave no reactivity.45 It was postulated that the pendant carboxylate leaving group hampers the approach of the anionic nucleophile due to charge–charge repulsion. This could be circumvented by using TMS-phthalimide as the nucleophile, whereby the carboxylate formed after ionization of 56 is presumably silylated to allow in situ nucleophilic addition to occur giving the desired AAA product in high yield and enantioselectivity.46 This reaction has been applied to the total synthesis of Tamiflu.46
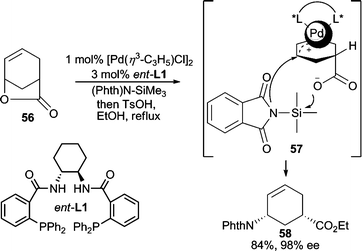 | ||
| Scheme 17 Ring-opening AAA with (Phth)N–SiMe3 as nucleophile. | ||
Desymmetrization of a meso-bis(carbamate) has been reported using the standard Trost ligand in excellent ee. Here, a tosylcarbamate functionality serves as both the nucleophile and the leaving group in the reaction.47,48 Vinyl aziridines49 have also been employed in Pd-catalyzed AAA reactions using acyl carbamates as nucleophiles to afford protected 1,2-diamines.
Acyclic carbonates participate in AAA reactions using a variety of imide nucleophiles in high regio- and stereoselectivities using Ir-catalysis.17,18,50 Generally high yields and ee values are obtained.
Heterocycles
Heterocycles stabilized by delocalization constitute another category of good nucleophiles for AAA reactions.1c Trost and Shi reported the first example of such a type51 by performing a Pd-catalyzed desymmetrization of meso-furan 59 with 6-chloropurine or 4-methoxypyrimidone as the nucleophile (Scheme 18). This methodology allows for the asymmetric preparation of nucleoside analogues after further manipulation of 61. Pyrroles6,52 (Scheme 3 and 19) and indoles53 have also been demonstrated in Pd-catalyzed AAA reactions. With vinyl aziridine electrophiles, a hydrogen bond between the pyrrole N–H and the ionized vinyl aziridine is believed to play a critical role in controlling the regioselectivity of the reaction. The AAA products of these pyrrole reactions are useful for the synthesis of a variety of pyrrole alkaloid natural products.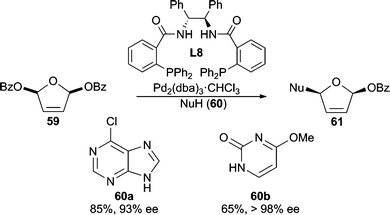 | ||
| Scheme 18 AAA for accessing nucleoside precursors. | ||
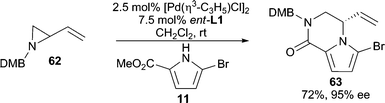 | ||
| Scheme 19 AAA with a substituted pyrrole. | ||
Oxygen nucleophiles
Stereogenic carbon atoms bearing oxygen as a heteroatom are present in many natural products and useful organic molecules. AAA with oxygen-based nucleophiles offers a significant opportunity to prepare such compounds in high enantiopurity. Furthermore, because these reactions typically operate under mild reaction conditions, elaborate coupling partners with a wide array of functional groups are typically tolerated in these reactions.Phenols
Phenols represent one of the best classes of oxygen nucleophiles for AAA reactions. They have been successfully utilized in a variety of allylic systems. When reacting 4-methoxyphenol with cyclohexenyl carbonate 64, high yield and enantioselectivity are obtained (Scheme 20).54 The product (65) can undergo an aromatic Claisen rearrangement to yield chiral phenol 66 with high chirality transfer. This serves as a useful procedure for accessing a stereogenic C–C bond via initial asymmetric C–O bond formation. Linear electrophiles have also been employed in this AAA/Claisen sequence.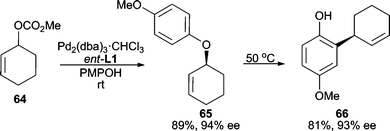 | ||
| Scheme 20 Stereoselective C–C bond formation through a Pd-catalyzed AAA/aromatic Claisen sequence. | ||
Due to the high functional group tolerance of Pd-catalyzed AAA reactions with phenol nucleophiles, this methodology can be a powerful tool for total synthesis. A Pd-catalyzed AAA reaction employing a highly substituted phenol nucleophile has been used for the total syntheses of (−)-galanthamine, (−)-codeine, and (−)-morphine.55
Unsymmetrical acyclic systems can also be employed in Pd-catalyzed AAA reactions with substituted phenols as nucleophiles using the Trost family of chiral ligands to afford products in good branched selectivities (91:9 to 98:2) and enantioselectivities (76–90% ee).56 This system was applied to the total synthesis of (−)-calanolides A and B, and the vitamin E nucleus. Furthermore, when using a chiral non-racemic allylic electrophile, a Pd-catalyzed AAA reaction proceeds with high catalyst control (19:1 dr)57 and has been applied to the total synthesis of deschlorocallipeltoside A.
Intramolecular AAA reactions of phenol nucleophiles can also be achieved in good yields and high enantioselectivities58 and have been applied to the total synthesis of (−)-siccanin.59 The addition of AcOH provides a dramatic improvement in enantioselectivity along with providing a reversal in the absolute stereochemistry of the product relative to when AcOH is absent. Moreover, the olefin geometry of the allylic carbonate influences the enantioselectivity. Use of the (E)-allyl carbonate provides the product in 84% ee whereas use of the (Z)-allyl carbonate affords the product in 97% ee. This difference has been rationalized through a Curtin–Hammett scenario based on differences in the way each geometrical isomer of the electrophile interacts with the chiral space induced around the catalyst by the ligand.60
Morita–Baylis–Hilman adducts can be deracemized by employing them as electrophiles in AAA reactions (Scheme 21).61,62 This dynamic kinetic asymmetric transformation (DYKAT) is achieved under Pd-catalysis using the Trost family of chiral ligands. This process has been successfully applied to the total syntheses of furaquinocins A, B and E.
 | ||
| Scheme 21 Deracemization of Morita–Baylis–Hilman adducts through Pd-catalyzed AAA reaction. | ||
In addition to Pd-catalyzed AAA reactions employing phenol nucleophiles, Ir- and Ru-catalyzed processes are known. Both alkyl and aryl acyclic allylic carbonates undergo AAA reactions with LiOAr nucleophiles using the [Ir(cod)Cl]2/chiral phosphoramidite system developed by Hartwig in high branched/linear selectivities (87:13 to 99:1) and in good enantioselectivities (72–97% ee).63 Electron deficient aryloxides failed to react under these conditions. A planar chiral Ru-complex was developed for AAA reactions between acyclic allylic chlorides and phenol nucleophiles in good regio- and enantioselectivities.64 X-Ray crystallography of the Ru-π-allyl complex formed from the reaction of cinnamyl chloride with 69 (Scheme 22) indicated that attack of the phenol on the Ru-π-allyl complex intermediate must occur with retention of configuration to form the product with the correct absolute stereochemistry observed in the reaction. Thus, this result suggests that attack of the nucleophile on the Ru-π-allyl intermediate (70) is an inner sphere process. In addition to the pregenerated planar chiral complex 69, a chiral Ru-catalyst can also be generated in situ. A decarboxylative AAA was reported using [CpRu(CH3CN)3PF6] in conjunction with a chiral diamine ligand with quantitative yield, good enantioselectivity, and high branched selectivity.65 A similar enantioselective decarboxylative process has also been reported with a Rh-catalyst with excellent regioselectivity but only 23% ee.66
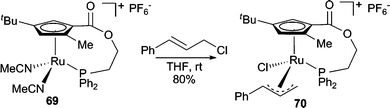 | ||
| Scheme 22 Reaction of a planar chiral Ru-complex with cinnamyl chloride. | ||
1,2-Diketone
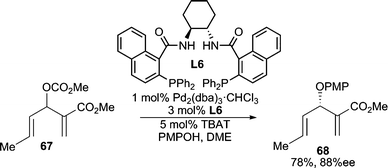
3-Substituted cyclic 1,2-diketones exist as a single tautomeric structure 71 (Scheme 23). This conjugated enol-like unit can serve as a useful nucleophile for AAA reactions.67 The O-alkylated products (73) are preferentially formed in good yields and high enantiomeric excesses with different ring sizes. These products are useful synthons for Lewis acid catalyzed Claisen rearrangements, which occur with high chirality transfer (ct) to give enantiomerically enriched C-alkylation products (74, 75). Linear acyclic allylic carbonates and vinyl epoxides can also be used as electrophiles in these AAA reactions. This method has been successfully applied to the total synthesis of (−)-terpestacin.68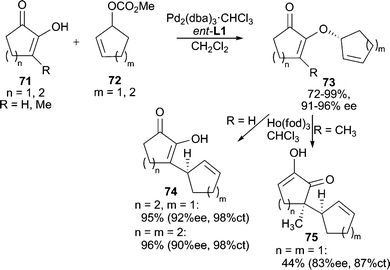 | ||
| Scheme 23 AAA reaction using diosphenol nucleophiles. | ||
Nitronates
In AAA reactions, nitronates are often known as nucleophiles for C-alkylation, generating nitroalkane functionalities.45a,69 By tuning steric and electronic factors on the nitronate, O-alkylation can also be achieved (Scheme 24). By substituting the nitronate with a sterically encumbering group such as isopropyl, and a conjugative stabilizing phenyl group (i.e.77), the nucleophilicity of the nitronate carbon is reduced whereas the nitronate oxygen atom becomes more nucleophilic.70 Following nucleophilic addition, fragmentation of 78 occurs to ultimately give enone 79 as the final product. Overall, this process allows for a formal oxidative desymmetrization of the meso-benzoate (76).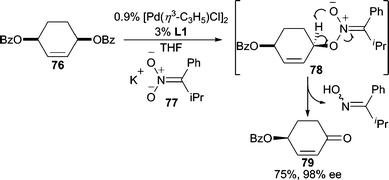 | ||
| Scheme 24 Nitronate nucleophiles. | ||
Hydroxylamines and oximes
Free hydroxylamines (also known as hydroxamic acids) and oximes may serve as either oxygen or nitrogen nucleophiles depending on the nature of the nucleophile. When the N-substituent is an electron-withdrawing group, nucleophilic attack by the oxygen moiety of the hydroxylamine becomes preferential. Efforts have been made in developing AAA reactions using such nucleophiles. Only an iridium/Pybox complex has been found successful for this operation.71 Oximes have also been investigated under the same reaction conditions, giving higher yields, b/l ratios, and enantioselectivities.32,72Carboxylates
Using carboxylates as oxygen nucleophiles is attractive since allylic esters often provide desired allylic alcohol functionalities after saponification. The problem stands, however, that the product allylic ester can also serve as an electrophile for AAA reactions. Thus, the reaction rate of the AAA with the starting ester must outcompete that of the product ester to minimize racemization of the product. To date, several examples have been reported using cyclic systems with carbonate leaving groups, due to their facile ionization, in conjunction with relatively electron-rich carboxylates as nucleophiles because of their reluctance to ionize. For example, methyl cycloheptenyl carbonate can be deracemized to form the resultant pivalate, isobutyrate, or propionate in excellent yields and enantioselectivities.73 In addition, sodium tiglate, an unsaturated carboxylate, is also a competent nucleophile in the reaction. Use of carboxylate nucleophiles has been applied to the total synthesis of (+)-phyllanthocin.74In an intriguing example employing racemic C2-symmetric allylic carbonate 80 (Scheme 25), stereoselective ionization followed by nucleophilic addition with benzoic acid leads initially to a monoalkylated allylic benzoate product, which then undergoes a second AAA reaction to furnish the bis(alkylation) product 81.75 Chemoselective deprotection of the remaining Troc-groups can be achieved with Zn metal in good yield.
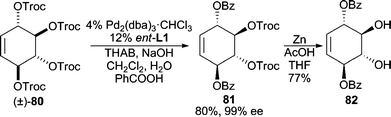 | ||
| Scheme 25 Double AAA reaction employing benzoate as nucleophile. | ||
Very recently, the planar chiral Ru-complex 69 (Scheme 22) was shown to be efficient in AAA reactions employing sodium carboxylate nucleophiles (Scheme 26).76 Gratifyingly, both aryl and alkyl allylic chlorides and aryl and alkyl carboxylate nucleophiles can be employed in this reaction with excellent results. Notably, allylic chlorides performed more efficiently than allylic bromides (reduced ee) or allylic carbonates (no reaction), and sodium carboxylates were more efficient relative to other alkali metal carboxylates (reduced yield and ee).
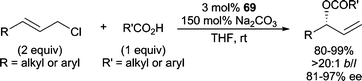 | ||
| Scheme 26 Ru-catalyzed AAA with sodium carboxylates. | ||
Hydrogen carbonate and carbonates
Alkali metal carbonates or bicarbonates are generally considered poor nucleophiles for allylic alkylation reactions, and therefore, are typically used as bases in such reactions.77 However, in AAA reactions, there have been reports of the use of these as nucleophiles to generate chiral allylic alcohols, thus functioning as convenient water surrogates. An early report uses vinyl epoxides as the electrophile with NaHCO3 as the nucleophile.78 The product formed in the reaction is a 5-membered cyclic carbonate, however, if BEt3 is employed as a cocatalyst (1 mol%) the vinylglycidol product can be isolated directly.Deracemization of cyclic and acyclic allylic carbonates using HCO3− as the nucleophile, which is generated in situ from saponification of the leaving group with water, results in formation of allylic alcohols in good yields and enantioselectivities (Scheme 27).79,80 In this process, the leaving group is hydrolyzed by water generating the HCO3− nucleophile. Nucleophilic attack of the π-allyl initially affords allylic hydrogen carbonate 86, which after loss of CO2 produces the observed allylic alcohol product. The possibility of water or hydroxide being the actual nucleophile in this process was excluded by control experiments.
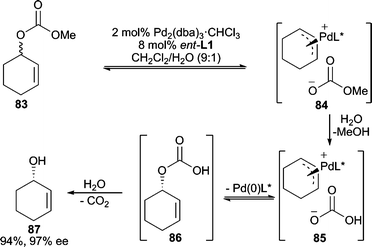 | ||
| Scheme 27 Deracemization of allylic carbonates by formal hydrolysis. | ||
Silanols
Similar to hydrogen carbonates, silanols or siloxides can also serve as hydroxide equivalents, forming chiral allylic alcohols in AAA reactions directly or silyl protected alcohols. This was first demonstrated by Trost and co-workers using vinyl epoxide electrophiles.81 Interestingly, the reaction regioselectivity could be controlled allowing access to either the branched or linear products. Carreira and co-workers reported the use of TESOK as a nucleophile in Ir-catalyzed AAA reactions employing acyclic allylic carbonate electrophiles.82 The allylic silyl ether products are formed in high yields, regio- and enantioselectivities and can be desilylated under mild conditions to obtain the corresponding allylic alcohols.Alcohols
Aliphatic alcohols and alkoxides are notoriously poor nucleophiles for AAA reactions, and when used in an alkali alkoxide form, elimination and deactivation of the metal catalyst can occur. Thus, activation of the reaction system toward alcohol attack or softening of the alkoxide nucleophile could potentially overcome these problems.As such, intramolecular AAA processes with neutral alcohol nucleophiles proceed efficiently, presumably because the close proximity of the hydroxyl group to the intermediate π-allyl complex facilitates the reaction. This has been exploited for tandem intermolecular Ru-catalyzed enyne cross-coupling followed by intramolecular AAA of the pendant alcohol giving tetrahydropyrans in high yields and enantioselectivities in an analogous manner to the amino version of this reaction given in Scheme 15.40,83 Another example was demonstrated in a recent synthesis of communiol A, where a catalyst controlled intramolecular AAA reaction was employed for the formation of the thermodynamically less stable 1,5-trans-tetrahydrofuran, which could then be elaborated to the natural product.84
Alkoxides can also be used if “softened” under appropriate conditions. An early example involves the use of a trialkylborane as an additive, which generates the borate species in situ (Scheme 28).85 In this example, the AAA reaction between vinyl epoxide 88 and PMBOH, BEt3 was used as the co-catalyst which can deliver the borate nucleophile via89 furnishing the branched product. Excellent yields and enantioselectivities are obtained in this reaction.
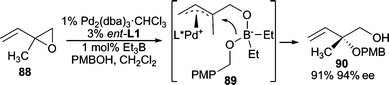 | ||
| Scheme 28 BEt3 co-catalyzed AAA reaction with aliphatic alcohols. | ||
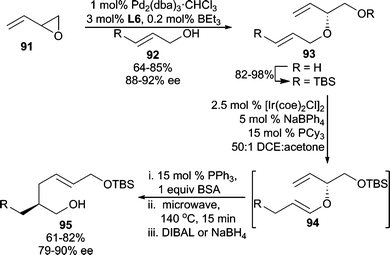
In an extension of this method, butadiene monoepoxide (91) was used as the electrophile in conjunction with an allylic alcohol nucleophile to access bis(allylic) ether 93 (Scheme 29).84 Subsequent alcohol protection and Ir-catalyzed allylic transposition generates 94in situ, which undergoes Claisen rearrangement upon heating affording α-chiral aldehydes. The aldehydes are reduced for isolation purposes to furnish alcohols (95) in good enantioselectivities for the overall reaction sequence. This olefin isomerization/Claisen process is known as the ICR.86
 | ||
| Scheme 29 AAA followed by ICR. | ||
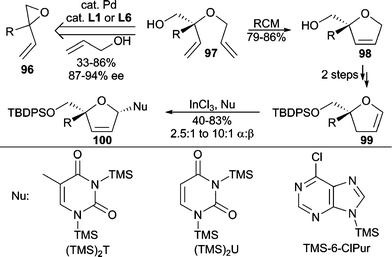
Important nucleoside analogues have been conveniently accessed through AAA chemistry by applying a tandem AAA/RCM sequence (Scheme 30).87 Bis(allyl) ethers (97) are prepared by AAA between allyl alcohol and vinyl epoxides (96) with good enantiocontrol. Dihydrofuran 100 is then readily prepared from 97 and is a useful precursor to densely functionalized nucleoside analogues.
 | ||
| Scheme 30 Tandem AAA/RCM en route to nucleoside analogues. | ||
Other metal alkoxides have also been studied for allylic alkylations. Most notable are Cu-alkoxides, which in the presence of a chiral Ir-catalyst undergo AAA reactions with acyclic allylic carbonate electrophiles in excellent yields, regio-, and enantioselectivities.88 Both primary and secondary alcohols work well, whereas tertiary alcohols afford products in low enantioselectivities. Additionally, zinc alkoxides have been shown to be effective in allylic etherification reactions; however, an asymmetric variant has not yet been reported.89
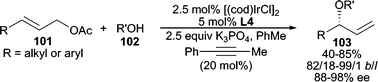
More recently, alcohols have been used directly in intermolecular allylic etherification. By utilizing K3PO4 as base, Ir-catalyzed AAA using alcohols is achieved with acyclic allylic acetates in good yields and selectivities (Scheme 31).90 Interestingly, 20 mol% of 1-phenyl-1-propyne was necessary to minimize double bond isomerization of the product. Both primary and secondary alcohols reacted well, whereas tertiary alcohols encountered lower reactivity yet maintained high enantioselectivity. Similarly, planar chiral Ru-complex 69 (Scheme 22) was also effective in the allylic etherification between allylic chlorides and MeOH or BnOH utilizing K2CO3 as the base.64
 | ||
| Scheme 31 Ir-catalyzed AAA reaction employing aliphatic alcohols. | ||
Sulfur nucleophiles
Chiral sulfur-containing compounds are highly useful intermediates in asymmetric organic synthesis. However, methods for asymmetric C–S bond construction are largely underdeveloped. Transition-metal-catalyzed AAA reactions offer a potential strategy for addressing this need. A variety of sulfur nucleophiles can be used, such as sulfones, thiocarboxylates, and thiols.Sulfones
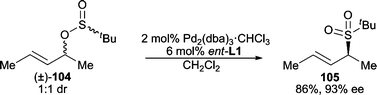
Sulfones are versatile intermediates in organic synthesis. The use of sulfones as nucleophiles in AAA reactions is successful for a variety of systems, including cyclic,91 acyclic,92 unsymmetrical,93 and gem-diacetate94 electrophiles.In addition to using an added external sulfone as the nucleophile, “internal nucleophiles” have also been used to form chiral sulfones through an enantioselective “rearrangement” from a racemic 1:1 diastereomeric mixture of allyl sulfinates (Scheme 32).95
 | ||
| Scheme 32 Deracemization of allylic sulfinates. | ||
Thiocarboxylates
Thiocarboxylates are another successful class of sulfur nucleophiles for AAA reactions, which lead to the formation of synthetically useful chiral allylic thiocarbamates. The Gais group has demonstrated that both cyclic and acyclic allylic carbonates can be attacked by thiocarboxylate anions to give satisfying reactivity and enantioselectivity (Scheme 33).96 Alternatively, chiral S-allylic thiocarbamates can be formed in the presence of a chiral catalyst via 1,3-rearrangement of racemic O-allylic thiocarbamates.97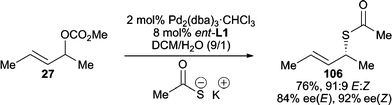 | ||
| Scheme 33 AAA reaction employing a thiocarboxylate. | ||
Thiols
In general, few reports of using thiol nucleophiles have been disclosed. The Gais group has studied the formation of chiral allylic sulfides using 2-pyrimidinethiol as the nucleophile with racemic allylic carbonate electrophiles.92,98Other nucleophiles
In addition to N, O and S, there have been reports of using other heteroatoms as nucleophiles in AAA reactions. Among them, the most studied are hydride nucleophiles. Such reactions are mostly categorized into another reaction type: asymmetric allylic reduction.99 Preliminary results have been published on the use of silicon100 and phosphine101 nucleophiles (Scheme 34), however, these have not yet been developed into general systems. Additionally, asymmetric borylation has been achieved by employing B2(pin)2 (111) under Cu-catalyzed conditions.102 While this reaction likely proceeds through an SN2′ pathway with L*Cu–B(pin) as the nucleophile rather than the standard AAA reaction mechanism, this formal AAA reaction allows access to enantiomerically enriched allylic boronates for use in allylation chemistry. Enantiospecific borylation of allylic103 and propargylic104 carbonates has also been reported.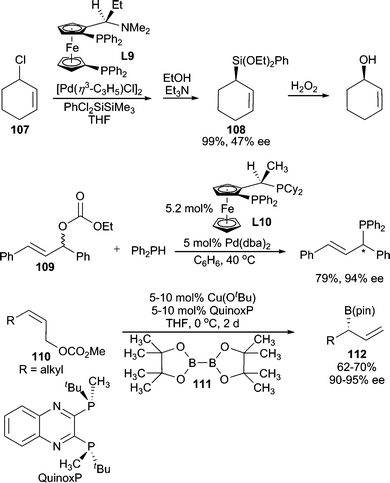 | ||
| Scheme 34 Si-, P-, and B-centered nucleophiles. | ||
Conclusions
Transition-metal-catalyzed AAA reactions continue to be a powerful synthetic tool in organic chemistry. Compared to stabilized carbon nucleophiles, the use of heteroatom nucleophiles in AAA reactions has been a constant challenge. To date, many efforts have been made in investigating the scope of heteroatom nucleophiles that can be successfully used in such transformations, and many successes have been achieved. A number of total syntheses have been published that take advantage of this powerful reaction class. The ability to tune the catalyst by varying the metal or ligand allows for chemo-, regio-, and stereoselective formation of a variety of bond types. Among the various heteroatom nucleophiles, nitrogen-centered nucleophiles offer the biggest variety and selectivity. Second are oxygen nucleophiles; although the activation of alcohol nucleophiles towards AAA remains a challenge, many encouraging results have been achieved. Other nucleophiles however are largely underdeveloped. Sulfur nucleophiles have found some exciting success. Preliminary results have been made with silicon and phosphine nucleophiles. Continual developments in the field related to nucleophile scope will empower this methodology to be an even more reliable tool for the organic chemist.Acknowledgements
We thank the NSF and NIH for funding of our research efforts in this area, and the Johnson Matthey Co. for the generous donation of precious metal salts. TZ acknowledges Roche for a graduate fellowship. JDS acknowledges the American Cancer Society for a post-doctoral fellowship.Notes and references
- Reviews: (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (b) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS.
- B. M. Trost and T. R. Verhoeven, J. Org. Chem., 1976, 41, 3215 CrossRef CAS.
- H. Matsushita and E.-I. Negishi, J. Chem. Soc., Chem. Commun., 1982, 160 RSC.
- B. M. Trost and M. L. Crawley, Chem.–Eur. J., 2004, 10, 2237 CrossRef CAS.
- (a) B. Crociani, F. Di Biana, A. Giovenco and T. Boschi, Inorg. Chim. Acta, 1987, 127, 169 CrossRef CAS; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545 CrossRef CAS.
- (a) B. M. Trost and G. Dong, J. Am. Chem. Soc., 2006, 128, 6054 CrossRef CAS; (b) B. M. Trost and G. Dong, Chem.–Eur. J., 2009, 15, 6910 CrossRef CAS.
- T. Nemoto, T. A. Masuda, T. Fukuyama and Y. Hamada, Org. Lett., 2005, 7, 4447 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Angew. Chem., Int. Ed. Engl., 1992, 31, 228 CrossRef.
- B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747 CrossRef CAS.
- C. P. Butts, E. Filali, G. C. Lloyd-Jones, P.-O. Norrby, D. A. Sale and Y. Schramm, J. Am. Chem. Soc., 2009, 131, 9945 CrossRef CAS.
- (a) T. Hayashi, A. Yamamoto, Y. Ito, E. Nishioka, H. Miura and K. Yanagi, J. Am. Chem. Soc., 1989, 111, 6301 CrossRef CAS; (b) M. Sawamura and Y. Ito, Chem. Rev., 1992, 92, 857 CrossRef CAS.
- (a) C. G. Frost, J. Howarth and J. M. J. Williams, Tetrahedron: Asymmetry, 1992, 3, 1089 CrossRef CAS; (b) C. G. Frost and J. M. J. Williams, Tetrahedron Lett., 1993, 34, 2015 CrossRef CAS; (c) C. G. Frost and J. M. J. Williams, Tetrahedron: Asymmetry, 1993, 4, 1785 CrossRef CAS; (d) P. von Matt and A. Pfaltz, Angew. Chem., Int. Ed. Engl., 1993, 32, 566 CrossRef; (e) J. Sprinz, M. Kiefler, G. Helmchen, M. Reggelin, G. Huttner, O. Walter and L. Zsolnai, Tetrahedron Lett., 1994, 35, 1523 CrossRef CAS; (f) J. M. Brown, D. I. Hulmes and P. J. Guiry, Tetrahedron, 1994, 50, 4493 CrossRef CAS; (g) A. Togni, U. Burckhardt, V. Gramlich, P. S. Pregosin and R. Salzmann, J. Am. Chem. Soc., 1996, 118, 1031 CrossRef CAS.
- Rh: (a) P. A. Evans, J. E. Robinson and K. K. Moffett, Org. Lett., 2001, 3, 3269 CrossRef CAS; (b) P. A. Evans and D. K. Leahy, J. Am. Chem. Soc., 2000, 122, 5012 CrossRef CAS; (c) P. A. Evans and D. K. Leahy, J. Am. Chem. Soc., 2002, 124, 7882 CrossRef CAS.
- B. M. Trost, P. L. Fraisse and Z. T. Ball, Angew. Chem., Int. Ed., 2002, 41, 1059 CrossRef CAS.
- Y. Yatsumonji, Y. Ishida, A. Tsubouchi and T. Takeda, Org. Lett., 2007, 9, 4603 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS.
- (a) R. Weihofen, O. Tverskoy and G. Helmchen, Angew. Chem., Int. Ed., 2006, 45, 5546 CrossRef CAS; (b) C. Gnamm, G. Franck, N. Miller, T. Stork, K. Brödner and G. Helmchen, Synthesis, 2008, 3331 CAS.
- M. J. Pouy, A. Leitner, D. J. Weix, S. Ueno and J. F. Hartwig, Org. Lett., 2007, 9, 3949 CrossRef CAS.
- T. Nagano and S. Kobayashi, J. Am. Chem. Soc., 2009, 131, 4200 CrossRef CAS.
- P. von Matt, O. Loiseleur, G. Koch, A. Pfaltz, C. Lefeber, T. Feucht and G. Helmchen, Tetrahedron: Asymmetry, 1994, 5, 573 CrossRef CAS.
- B. M. Trost, T. L. Calkins, C. Oertelt and J. Zambrano, Tetrahedron Lett., 1998, 39, 1713 CrossRef CAS.
- T. Hayashi and K. Kishi, Tetrahedron Lett., 1990, 31, 1743 CrossRef CAS.
- L.-L. You, X.-Z. Zhu, Y.-L. Luo, X.-L. Hou and Y.-M. Dai, J. Am. Chem. Soc., 2001, 123, 7471 CrossRef CAS.
- T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164 CrossRef CAS.
- (a) C. Welter, O. Koch, G. Lipowsky and G. Helmchen, Chem. Commun., 2004, 896 RSC; (b) C. Welter, A. Dahnz, B. Brunner, S. Streiff, P. Dubon and G. Helmchen, Org. Lett., 2005, 7, 1239 CrossRef CAS; (c) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675 RSC; (d) C. Gnamm, C. Krauter, K. Brödner and G. Helmchen, Chem.–Eur. J., 2009, 15, 2050 CrossRef CAS; (e) C. Gnamm, K. Brödner, C. Krauter and G. Helmchen, Chem.–Eur. J., 2009, 15, 10514 CrossRef CAS.
- G. Lipowsky and G. Helmchen, Chem. Commun., 2004, 116 RSC.
- Y. Matsushima, K. Onitsuka, T. Kondo, T. Mitsudo and S. Takahashi, J. Am. Chem. Soc., 2001, 123, 10405 CrossRef CAS.
- Y. Dong, P. Teesdale-Spittle and J. O. Hoberg, Tetrahedron Lett., 2005, 46, 353 CrossRef CAS.
- T. Nemoto, S. Tamura, T. Sakamoto and Y. Hamada, Tetrahedron: Asymmetry, 2008, 19, 1751 CrossRef CAS.
- (a) C. Shu, A. Leitner and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4797 CrossRef CAS; (b) A. Leitner, C. Shu and J. F. Hartwig, Org. Lett., 2005, 7, 1093 CrossRef CAS; (c) S. Shekhar, B. Trantow, A. Leitner and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 11770 CrossRef CAS; (d) D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 11680 CrossRef CAS.
- Y. Yamashita, A. Gopalarathnam and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7508 CrossRef CAS.
- H. Miyabe, A. Matsumura, K. Moriyama and Y. Takemoto, Org. Lett., 2004, 6, 4631 CrossRef CAS.
- (a) B. M. Trost and D. R. Fandrick, J. Am. Chem. Soc., 2003, 125, 11836 CrossRef CAS; (b) B. M. Trost and D. R. Fandrick, Org. Lett., 2005, 7, 823 CrossRef CAS.
- C. Larksarp and H. Alper, J. Am. Chem. Soc., 1997, 119, 3709 CrossRef CAS.
- B. M. Trost and S. Pulley, Tetrahedron Lett., 1995, 36, 8737 CrossRef CAS.
- B. M. Trost, J. Dudash, Jr. and E. J. Hembre, Chem.–Eur. J., 2001, 7, 1619 CrossRef CAS.
- B. M. Trost and J. D. Oslob, J. Am. Chem. Soc., 1999, 121, 3057 CrossRef CAS.
- R. Stragies and S. Blechert, J. Am. Chem. Soc., 2000, 122, 9584 CrossRef CAS.
- H. Tye, D. Smyth, C. Eldred and M. Wills, Chem. Commun., 1997, 1053 RSC.
- B. M. Trost and M. R. Machacek, Angew. Chem., Int. Ed., 2002, 41, 4693 CrossRef CAS.
- B. M. Trost, S. Malhotra, D. E. Olson, A. Maruniak and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 4190 CrossRef CAS.
- B. M. Trost and R. C. Bunt, J. Am. Chem. Soc., 1994, 116, 4089 CrossRef CAS.
- (a) B. M. Trost, R. C. Bunt, R. C. Lemoine and T. L. Calkins, J. Am. Chem. Soc., 2000, 122, 5968 CrossRef CAS; (b) B. M. Trost, A. C. Krueger, R. C. Bunt and J. Zambrano, J. Am. Chem. Soc., 1996, 118, 6520 CrossRef CAS; (c) B. M. Trost, C. Jiang and K. Hammer, Synthesis, 2005, 3335 CrossRef CAS; (d) B. M. Trost, D. B. Horne and M. J. Woltering, Angew. Chem., Int. Ed., 2003, 42, 5987 CrossRef CAS; (e) B. M. Trost, D. B. Horne and M. J. Woltering, Chem.–Eur. J., 2006, 12, 6607 CrossRef CAS.
- (a) B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931 CrossRef CAS; (b) B. M. Trost, A. Aponick and B. N. Stanzl, Chem.–Eur. J., 2007, 13, 9547 CrossRef CAS; (c) B. M. Trost and B. M. O'Boyle, Org. Lett., 2008, 10, 1369 CrossRef CAS.
- (a) B. M. Trost and J.-P. Surivet, Angew. Chem., Int. Ed., 2000, 39, 3122 CrossRef CAS; (b) B. M. Trost and R. C. Bunt, J. Am. Chem. Soc., 1996, 118, 235 CrossRef CAS.
- B. M. Trost and T. Zhang, Angew. Chem., Int. Ed., 2008, 47, 3759 CrossRef CAS.
- (a) B. M. Trost and D. E. Patterson, J. Org. Chem., 1998, 63, 1339 CrossRef CAS; (b) B. M. Trost, Z. Pan, J. Zambrano and C. Kujat, Angew. Chem., Int. Ed., 2002, 41, 4691 CrossRef CAS.
- B. M. Trost and D. E. Patterson, Chem.–Eur. J., 1999, 5, 3279 CrossRef CAS.
- B. M. Trost, D. R. Fandrick, T. Brodmann and D. T. Stiles, Angew. Chem., Int. Ed., 2007, 46, 6123 CrossRef CAS.
- (a) S. Spiess, C. Berthold, R. Weihofen and G. Helmchen, Org. Biomol. Chem., 2007, 5, 2357 RSC; (b) O. V. Singh and H. Han, Tetrahedron Lett., 2007, 48, 7094 CrossRef CAS.
- B. M. Trost and Z. Shi, J. Am. Chem. Soc., 1996, 118, 3037 CrossRef CAS.
- B. M. Trost and G. Dong, Org. Lett., 2007, 9, 2357 CrossRef CAS.
- B. M. Trost, M. J. Krische, V. Berl and E. M. Grenzer, Org. Lett., 2002, 4, 2005 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 815 CrossRef CAS.
- (a) B. M. Trost and W. Tang, J. Am. Chem. Soc., 2002, 124, 14542 CrossRef CAS; (b) B. M. Trost and W. Tang, Angew. Chem., Int. Ed., 2002, 41, 2795 CrossRef CAS; (c) B. M. Trost, W. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 9074 CrossRef CAS.
- (a) B. M. Trost and J. L. Gunzner, J. Am. Chem. Soc., 2001, 123, 9449 CrossRef CAS; (b) B. M. Trost, J. L. Gunzner, O. Dirat and Y. H. Rhee, J. Am. Chem. Soc., 2002, 124, 10397.
- B. M. Trost, H. C. Shen, L. Dong and J.-P. Surivet, J. Am. Chem. Soc., 2003, 125, 9276 CrossRef CAS.
- (a) B. M. Trost, H. C. Shen and J.-P. Surivet, Angew. Chem., Int. Ed., 2003, 42, 3943 CrossRef CAS; (b) B. M. Trost and H. C. Shen, J. Am. Chem. Soc., 2004, 126, 12565 CrossRef CAS.
- B. M. Trost, H. C. Shen, L. Dong and J.-P. Surivet, J. Am. Chem. Soc., 2004, 126, 11966 CrossRef CAS.
- B. M. Trost, O. R. Thiel and H.-C. Tsui, J. Am. Chem. Soc., 2003, 125, 13155 CrossRef CAS.
- B. M. Trost and M. K. Brennan, Org. Lett., 2007, 9, 3961 CrossRef CAS.
- F. Lopez, T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 3426 CrossRef CAS.
- K. Onitsuka, H. Okuda and H. Sasai, Angew. Chem., Int. Ed., 2008, 47, 1454 CrossRef CAS.
- M. Austeri, D. Linder and J. Lacour, Chem.–Eur. J., 2008, 14, 5737 CrossRef CAS.
- G. Consiglio, M. Scalone and F. Rama, J. Mol. Catal., 1989, 50, L11 CrossRef CAS.
- B. M. Trost and G. M. Schroeder, J. Am. Chem. Soc., 2000, 122, 3785 CrossRef CAS.
- B. M. Trost, G. Dong and J. A. Vance, J. Am. Chem. Soc., 2007, 129, 4540 CrossRef CAS.
- H. Rieck and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1995, 34, 2687 CrossRef CAS.
- B. M. Trost, J. Richardson and K. Yong, J. Am. Chem. Soc., 2006, 128, 2540 CrossRef CAS.
- H. Miyabe, K. Yoshida, M. Yamauchi and Y. Takemoto, J. Org. Chem., 2005, 70, 2148 CrossRef CAS.
- H. Miyabe, A. Matsumura, K. Yoshida and Y. Takemoto, Tetrahedron, 2009, 65, 4464 CrossRef CAS.
- B. M. Trost and M. G. Organ, J. Am. Chem. Soc., 1994, 116, 10320 CrossRef CAS.
- B. M. Trost and Y. Kondo, Tetrahedron Lett., 1991, 32, 1613 CrossRef CAS.
- B. M. Trost, D. E. Patterson and E. J. Hembre, J. Am. Chem. Soc., 1999, 121, 10834 CrossRef CAS.
- N. Kanbayashi and K. Onitsuka, J. Am. Chem. Soc., 2010, 132, 1206 CrossRef CAS.
- A.-A. G. Shaikh and S. Swairam, Chem. Rev., 1996, 96, 951 CrossRef CAS.
- B. M. Trost and E. J. McEachern, J. Am. Chem. Soc., 1999, 121, 8649 CrossRef CAS.
- B. J. Lussem and H.-J. Gais, J. Am. Chem. Soc., 2003, 125, 6066 CrossRef.
- H.-J. Gais, O. Bondarev and R. Hetzer, Tetrahedron Lett., 2005, 46, 6279 CrossRef CAS.
- (a) B. M. Trost, N. Ito and P. D. Greenspan, Tetrahedron Lett., 1993, 34, 1421 CrossRef CAS; (b) B. M. Trost, P. D. Greenspan, H. Geissler, J. H. Kim and N. Greeves, Angew. Chem., Int. Ed. Engl., 1994, 33, 2182 CrossRef.
- I. Lyothier, C. Defieber and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 6204 CrossRef CAS.
- B. M. Trost, M. R. Machacek and B. D. Faulk, J. Am. Chem. Soc., 2006, 128, 6745 CrossRef CAS.
- B. M. Trost and T. Zhang, Org. Lett., 2006, 8, 6007 CrossRef CAS.
- B. M. Trost, E. J. McEachern and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 12702 CrossRef CAS.
- (a) S. G. Nelson, C. J. Bugard and K. Wang, J. Am. Chem. Soc., 2003, 125, 13000 CrossRef CAS; (b) S. G. Nelson and K. Wang, J. Am. Chem. Soc., 2006, 128, 4232 CrossRef CAS.
- B. M. Trost, B. S. Brown, E. J. McEachern and O. Kuhn, Chem.–Eur. J., 2003, 9, 4442 CrossRef CAS.
- C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794 CrossRef CAS.
- H. Kim and C. Lee, Org. Lett., 2002, 4, 4369 CrossRef CAS.
- S. Ueno and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 1928 CrossRef CAS.
- B. M. Trost, M. G. Organ and G. A. O'Doherty, J. Am. Chem. Soc., 1995, 117, 9662 CrossRef CAS.
- H.-J. Gais, T. Jagusch, N. Spalthoff, F. Gerhards, M. Frank and G. Raabe, Chem.–Eur. J., 2003, 9, 4202 CrossRef CAS.
- H. Eichelmann and H.-J. Gais, Tetrahedron: Asymmetry, 1995, 6, 643 CrossRef CAS.
- B. M. Trost, M. L. Crawley and C. B. Lee, Chem.–Eur. J., 2006, 12, 2171 CrossRef CAS.
- T. Jagusch, H.-J. Gais and O. Bondarev, J. Org. Chem., 2004, 69, 2731 CrossRef CAS.
- B. J. Lussem and H.-J. Gais, J. Org. Chem., 2004, 69, 4041 CrossRef.
- (a) A. Bohme and H.-J. Gais, Tetrahedron: Asymmetry, 1999, 10, 2511 CrossRef; (b) H.-J. Gais and A. Bohme, J. Org. Chem., 2002, 67, 1153 CrossRef CAS.
- H.-J. Gais, N. Spalthoff, T. Jagusch, M. Frank and G. Raabe, Tetrahedron Lett., 2000, 41, 3809 CrossRef CAS.
- T. Hayashi, H. Iwamura, M. Naito, Y. Matsumoto and Y. Uozumi, J. Am. Chem. Soc., 1994, 116, 775 CrossRef CAS.
- T. Hayashi, A. Ohno, S.-J. Lu, Y. Matsumoto, E. Fukuyo and K. Yanagi, J. Am. Chem. Soc., 1994, 116, 4221 CrossRef CAS.
- P. Butti, R. Rochat, A. D. Sadow and A. Togni, Angew. Chem., Int. Ed., 2008, 47, 4878 CrossRef CAS.
- H. Ito, S. Ito, Y. Sasaki, K. Matsuura and M. Sawamura, J. Am. Chem. Soc., 2007, 129, 14856 CrossRef CAS.
- H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034 CrossRef CAS.
- H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |