A versatile, solvent-free methodology for the functionalisation of carbon nanotubes†
Robert
Menzel
a,
Michael Q.
Tran
b,
Angelika
Menner
b,
Christopher W. M.
Kay
c,
Alexander
Bismarck
b and
Milo S. P.
Shaffer
*a
aDepartment of Chemistry, Imperial College, London, UK SW7 2AZ. E-mail: m.shaffer@imperial.ac.uk; Fax: +44(0)2075945801; Tel: +44(0)2075945825
bDepartment of Chemical Engineering, Imperial College, London, UK SW7 2AZ
cInstitute of Structural and Molecular Biology, University College London, Gower Street, London, UK WC1E 6BT
First published on 11th August 2010
Abstract
High temperature activation of carbon nanotubes (CNTs) provides a new and highly versatile functionalisation strategy. The reaction allows the attachment of a wide variety of functional species onto the nanotube surface at grafting ratios between 1–8 wt%, whilst maintaining the intrinsic properties of the untreated materials. The underlying, radical-based, reaction mechanism has been established by quenching experiments and EPR studies. The distribution of the functionalised sites has been investigated at the microscopic scale using tagging reactions. The grafted products have been characterized by electron microscopy, thermal analysis (TGA), Raman spectroscopy, and inverse gas chromatography (IGC). The change in the CNT surface properties after grafting has been quantified in terms of dispersive and specific surface energies, and altered dispersibilities in a broad range of solvents. It is possible to carry out the reaction using gas phase reagents, providing a clean, efficient, and scalable methodology, relevant to a diverse range of applications.
Introduction
It has long been known that heating of graphitic materials under inert atmospheres to around 1000 °C leads to the decomposition of the surface oxides.1 Depending on the temperature at which the material is re-exposed to air, the surface oxides subsequently reform as either acidic or basic surface groups.2,3 The mechanism has not been elucidated, but these literature findings hint at the formation of active radicals on the graphitic surface, during high temperature treatment, which later trap oxygen. On that basis, we have investigated the hypothesis that the thermally-activated carbons ought to bind covalently, not just oxygen, but a wide variety of functional molecules, including polymerisable monomers. In particular, we have demonstrated the effect using carbon nanotubes (CNTs), for which new, more practical, functionalisation strategies are urgently sought. Due to their exceptional structural, electrical, and mechanical properties,4,5 CNTs are relevant to a wide range of applications, including engineering,6 bionanotechnology,7 and catalysis.8,9 Chemical modification is frequently required, in order to compatibilise the nanotubes with a particular environment or to attach functional components.10–12 For these reasons, strategies for the covalent functionalisation of CNTs have been intensively explored in recent years;12 numerous wet-chemical reactions are available, allowing the introduction of a wide range of functional surface groups.11,13 Examples include the chemical derivatisation of oxygen-containing functional groups at defect sites, e.g. through classical amidation or esterfication reactions,14 and direct functionalisation of the graphitic CNT sidewalls, such as by reactions with nitrenes,15 carbenes16 and diazonium salts.17 Since covalent modification of the CNT structure damages the intrinsic properties of interest, it is often desirable to attach macromolecular species that multiply the number of functional groups attached per reactive site on the nanotube surface.18 Free radical reactions are a common approach to nanotube functionalisation and macromolecule grafting, specifically; they divide into two general strategies.19 In the “grafting-to” approach, free radicals are generated in the supporting solvent and subsequently terminated on the CNT surface; examples include the reaction of CNTs with pre-synthesised, azide- or nitroxyl-terminated macromolecules,20,21 or photolysable organic iodides.22 The “grafting-from” approach involves the polymerisation of monomers from traditional initiating species that have been first immobilised on the nanotube surface.11 Living radical polymerization methods, including atom transfer radical polymerization,23 nitroxide-mediated polymerization,24 and reversible addition-fragmentation chain transfer polymerization,25 have been frequently employed for this purpose due to their tolerance of a wide variety of functional groups.These conventional, wet-chemical, CNT derivatisation reactions are typically time-consuming and inconvenient, frequently requiring multiple reaction steps and/or lengthy filtration/centrifugation procedures for purification. In addition, they typically create large volumes of liquid waste, consisting of toxic organic solvent or corrosive mineral acids which can significantly limit the economical viability of these approaches. There is, therefore, a need to develop simpler, solvent-free methodologies. Although several, dry functionalisation methods have been reported, including direct fluorination,26 thermal oxidation,27 and plasma treatments,28 unfortunately, these reactions often lead to significant degradation of the graphitic framework and intrinsic CNT properties, and suffer from poor reproducibility and versatility. In this edge article, we present an alternative, thermochemical functionalisation approach, extending an existing method developed to alter the surface oxide chemistry of carbon materials, including carbon fibres29 and CNTs.27
Experimental
CNTs were synthesised in our laboratory (in-house CNTs) employing standard CVD-growth conditions yielding mats of relatively straight and aligned, large multi-wall carbon nanotubes (outer diameter 80–100 nm, length of a few hundred micrometres).30 Commercial, CVD-grown CNTs were obtained from Arkema SA (Lacq-Mourenx, France) and Nanocyl SA (Sambreville, Belgium) and consisted of aggregates of entangled CNTs with outer diameters of around 10–20 nm and lengths at least a few micrometres. Prior to the thermochemical treatment, the as-received or as-synthesized CNTs were pre-oxidised by heating in air (670 °C, cycles of 6 × 5 min) in order break-up the entangled CNT agglomerates and introduce additional oxygen-containing functional groups onto the CNT surfaces.27 These pre-oxidised CNTs are referred to as “parent” CNTs in this article.The grafting was carried out in a custom-made setup consisting of a 30 mm diameter quartz tube attached to a sample flask (Fig. 1). In order to work under air-free conditions, the setup was connected to an inert gas source or a vacuum system. 100 mg CNTs were heated to 1000 °C under oxygen-free nitrogen or vacuum (5 × 10−4 mbar) at 15 K min−1 in a conventional three-zone tube furnace (PTF 12/38/500, Lenton Ltd, UK) and held at temperature for 2 h. In a second step, the quartz tube was slowly removed from the heating zone and allowed to cool to room temperature. The CNTs were transferred to the round bottom flask by gravity. 3 mL of the reactant (Fig. 1) were injected either directly into the flask containing the thermally-activated sample (liquid-phase setup) or into an empty reservoir attached to the sample flask (gas-phase setup). After leaving the sample-monomer mixture under an inert atmosphere or vacuum for 12 h, non-reacted reactant was removed via filtration. Prior to characterisation, the product was thoroughly washed three times in order to remove any physisorbed reactants. Each washing step consisted of bath sonication for 5 min in 50 mL of the washing agent, filtration and rinsing with 3 × 50 mL. For comparative reasons, these washing steps were also applied to the products synthesized using the gas-phase setup. The washing procedure was a requirement for the detailed fundamental study in this paper; for application, simple removal and recovery of excess reactant via evaporation under vacuum, without using any solvents, should be sufficient.
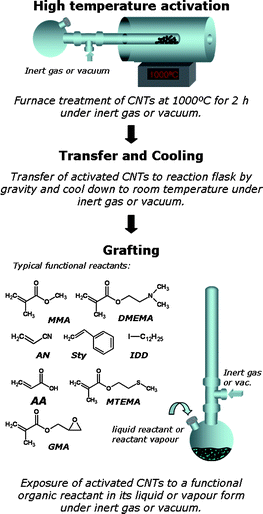 | ||
| Fig. 1 Schematic overview of the thermochemical grafting of carbon nanotubes with functional organic reactants. Refer to the ESI† (Table S1) for further details about the organic reactants used. | ||
Further experimental details can be found in the ESI.†
Results and discussion
As an illustration, commercial CNTs were high-temperature activated and treated with lauryl methacrylate (LMA) in oxygen-free nitrogen using the liquid-phase experimental setup followed by washing with toluene. Thermogravimetric analysis (TGA) of the LMA-treated sample in air confirmed successful grafting (Fig. 2(a)).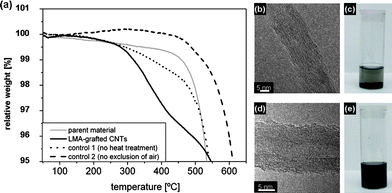 | ||
| Fig. 2 Characterization of CNTs grafted with lauryl methacrylate (LMA): TGA weight loss profiles of LMA-grafted CNTs and corresponding control samples (a); HRTEM images of a parent CNT (b) and LMA-grafted CNT (d); dispersion of parent CNTs (c) and LMA-grafted CNTs (e) in butyl acetate. | ||
Prior to the combustion of the CNTs at around 600 °C (only the onsets are shown in Fig. 2(a), see ESI for full TGA curves†), a small but distinct weight loss was observed at around 355 °C (Fig. 2(a)), associated with a clear peak in the derivative curve and indicating the combustion of the grafted organics. Two control experiments were carried out under identical treatment conditions. For control experiment 1, the parent CNTs were mixed with the LMA monomer under inert gas, omitting the high-temperature treatment; for control experiment 2, the heat-treated CNTs were mixed with LMA only after exposure to air for 1 h. The products underwent the same washing procedure as applied for the LMA-grafted CNTs. The first control showed a very small, broad weight-loss, with no peak in the derivative; the feature can be attributed to modest physisorption on the heterogeneous CNT surface,31 caused either by the adsorption of LMA monomer in slit pores or on iron impurities inherently present in these CNTs. In the second control, the slightly rising profile and increased thermal stability of the CNTs is consistent with the presence of stable basic surface oxides; it has been shown previously that heat-activated carbons exposed to air at room temperature form stable basic and ether-type oxygen-containing surface groups3,32 that decompose at elevated temperatures following take-up of additional oxygen,27,33 explaining the initial weight gain observed. The consistently different weight loss profiles (Fig. 2(a)), therefore, confirm that high-temperature activation and the exclusion of air are prerequisites for successful LMA grafting. From the TGA weight loss profile (Fig. 2(a)), the LMA grafting ratio (i.e. the weight of the chemisorbed organic monomer relative to the total weight of the product) can be estimated to be 3.0 wt% (see also Fig. 1 in the ESI) which roughly equates to a CNT surface coverage of around 20%. After three repeats, the reproducibility of the LMA-grafting reaction was estimated to be Δ = (2.8 ± 0.8) wt%. Due to the rough surface and beam sensitivity of organic molecules, it is not surprising that the grafted monomer cannot be resolved in HRTEM images (Fig. 2(d)). However, a clear change in CNT surface character after grafting was detected via inverse gas chromatography; this sensitive analytical technique allows the determination of dispersive surface energies, γd, associated purely with London forces, as well as more chemically-specific surface characteristics. The specific interactions can be quantified in various ways, but here are reported in terms of acceptor and donor numbers, KA and KD, reflecting the relative ability of the (modified) CNT surface to undergo electron accepting and donating interactions.31 After LMA-grafting, the dispersive surface energy, γd, of the CNTs is significantly reduced from (113 ± 2) mJ m−2 to (87 ± 2) mJ m−2, which is consistent with the occupation or replacement of high-energy sites on the CNT surface with organic monomers.34 In addition, the KD/KA ratio increased from 2.3 ± 0.1 to 3.1 ± 0.1, indicating a more pronounced electron-donating surface character due the attachment of alkyl chains onto the CNT surface. These changes in dispersive and specific surface character are sufficient to alter the dispersion behavior, markedly. Compared to the parent material, the dispersibility of the LMA-grafted CNTs in butyl acetate increased by a factor of ten, from 3 to 35 mg L−1 (Fig. 2(c) and (e)), but substantially decreased by a factor of five in ethanol (Table 1). These differences in dispersibility were even more pronounced when the LMA-grafted product was compared to the ‘air-exposed’ control sample (Table 1) which has a relatively less polar surface than the parent material.
| Grafted compound (acronym) | Grafted compound (chemical name) | Setup | Grafting ratio Δ [wt%] | Monomeric units | Disp. surface energy γd/mJ m−2 | K D/KA | Concentration of dispersed CNTs/mg L−1 | ||
|---|---|---|---|---|---|---|---|---|---|
| Butyl acetate | Ethanol | Water (pH 4) | |||||||
| Parent CNTs | n/a | n/a | n/a | n/a | 113 | 2.3 | 3.2 | 14.0 | 0.1 |
| Air-exposed CNTs | n/a | n/a | n/a | n/a | 110 | 2.4 | 0.3 | 2.3 | 0.1 |
| Sty | Styrene | LP | 0.5 | 2 | 108 | 2.5 | |||
| IDD | Iodododecane | LP | 0.9 | 1 | 101 | 2.7 | |||
| MMA | Methyl methacrylate | GP | 2.3 | 10 | 83 | 3.0 | |||
| MMA | Methyl methacrylate | LP | 2.5 | 12 | 81 | 3.2 | 30.7 | 1.6 | 0.2 |
| LMA | Lauryl methacrylate | LP | 2.8 | 6 | 87 | 3.1 | 35.6 | 2.6 | 0.2 |
| GMA | Glycidyl methacrylate | LP | 3.0 | 6 | 84 | 2.6 | |||
| DMAEMA | 2-(Dimethylamino)ethyl methacrylate | LP | 5.2 | 16 | 85 | 2.9 | 2.1 | 5.3 | 8.7 |
| AN | Acrylonitrile | GP | 7.2 | 68 | 81 | 2.6 | 42 | 21 | 0.1 |
| AN | Acrylonitrile | LP | 7.3 | 68 | 77 | 2.5 | |||
| AA | Acrylic acid | LP | 7.9 | 54 | 60 | 1.9 | |||
The two control experiments in Fig. 2(a) show that reactive sites are generated during the heat activation step (unlike control experiment 1) but are quenched when exposed to air (control experiment 2). The nature of these reactive sites was further studied using EPR spectroscopy, which allows the detection of species with unpaired electrons. The EPR spectrum of the heat-treated commercial CNTs in vacuum exhibited a relatively narrow signal (g-factor of around 2.01) at a measurement temperature of 6 K, a temperature regime where the EPR spectra of conducting materials, such as CNTs, are expected to be dominated by localized spins, such as radicals and paramagnetic ions, rather than by the spin resonance of their delocalised conduction electrons.35–38 The EPR signal observed was quenched when the CNTs were exposed to air. These observations support the hypothesis that reactive radicals form on the CNT surface, associated with the desorption of surface oxides at high temperatures.
The surface radical concentration was estimated, using galvinoxyl, an air-stable radical with a characteristic UV-Vis absorption band at 434 nm. The absorption intensity of galvinoxyl in toluene only marginally changed when mixed with as-received CNTs but significantly decreased when added to the heat-activated CNTs (Fig. 3(b)), presumably due to the binding of galvinoxyl radicals from solution to the radicals on the CNT surface. By assuming that one galvinoxyl radical is quenched by one surface radical, the concentration of the active sites on the CNT can be calculated as 31 μmol per gram of CNTs. In a second, independent, quenching experiment, thermally-activated CNTs were reacted with iodododecane resulting in the grafting ratio of 0.9 wt% (Fig. 5). Again assuming a stoichiometric reaction, this grafting ratio corresponds to a radical concentration of 50 μmol g−1. The two independent quenching experiments indicate very similar surface radical concentrations, particularly given the difference in steric bulk; for the remainder of this article, we will use an average value of 40 μmol g−1 as an estimate for the concentration of grafting sites on the CNT surface. The grafting site concentration is significantly lower than the surface concentration of oxygen-containing groups on the parent CNTs, which was determined to be about 150 μmol g−1 by Böhm's titration with NaOH. This difference suggests that only certain types of surface oxides are precursors for the radicals, whilst the majority desorb in a heterolytic fashion or undergo migration and restructuring processes during the high temperature treatment.39,40 At a grafting ratio of 2.8 wt% LMA, i.e. 110 μmol LMA per gram CNT, there are about three times more monomeric units than grafting sites present on the CNT surface, implying that radical polymerization of the methacrylate has been initiated by the radicals on the CNT surface (i.e. a “grafting from” mechanism).11 Termination of the polymerization process may occur either through trace impurities in the reaction system, or via recombination of the propagating chain with a second radical site, resulting in oligomer loops on the CNT surface (Fig. 4). The latter option is likely to be favoured kinetically, leading to the low grafted molecular weight observed. On this basis, the covalently-bound LMA oligomers can be estimated to consist of six monomer repeats.
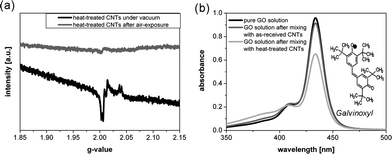 | ||
| Fig. 3 (a) EPR spectra of heat-treated Arkema CNTs in vacuum and after air exposure for 1 h, recorded at 6 K (the corresponding EPR spectra at room temperature are available in the ESI†); (b) UV-Vis spectra of a pure galvinoxyl solution in toluene after mixing with heat-activated CNTs and untreated CNTs under vacuum, respectively. | ||
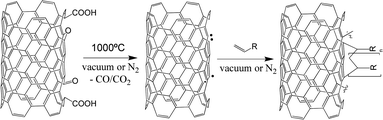 | ||
| Fig. 4 Proposed mechanism for the thermochemical activation and grafting of CNTs. | ||
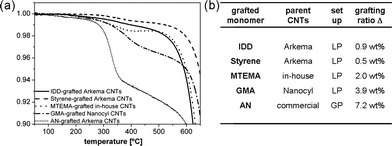 | ||
| Fig. 5 Versatility of the thermochemical grafting approach: (a) TGA weight loss profiles, and (b) grafting ratios for commercial and in-house grown CNTs grafted with various organic compounds. For acronyms and structures of the grafted compounds see Fig. 1 and ESI† (Table S1). | ||
The proposed grafting mechanism implies that the generation of the reactive sites on the CNT surface does not cause any significant additional damage to the graphitic network beyond the original oxidation; this assumption is confirmed by Raman measurements, which yield similar IG/ID ratios for the parent (0.85 ± 0.7) and LMA-grafted (0.81 ± 0.6) materials.
The underlying radical mechanism of the grafting reaction suggests that the thermochemical treatment approach should be a versatile methodology for the surface modification of CNTs. The generality was, therefore, successfully tested using CNTs of different dimensions and morphologies, and various reactants capable of reacting with radicals, including methacrylates, styrenes, and organic iodides (Fig. 5 and Table 1). Whilst the degree of functionalisation depends on the type of monomer and nanotube, the same reaction behaviour is observed in all cases. The grafting ratios and average oligomer chain lengths reached values up to 8 wt% and 70 monomeric units, respectively, for the most reactive compounds (Fig. 5; Table 1).
One particularly important question, often ignored in CNT chemistry, is how the reactive sites are distributed within the sample; this issue was probed using a tagging reaction. Similar approaches have been used previously for conventional wet-chemistry.41 In-house produced CNTs were grafted with both LMA and 2-(methylthio)ethyl methacrylate (MTEMA). By tagging the sulfur groups in MTEMA with gold colloids, the markedly different surface character of the two modified samples was confirmed (Fig. 6). While scanning electron microscopy images show binding of the gold particles to the MTEMA-grafted CNTs, no tagging of the LMA-grafted control sample is observed. Although a one-to-one correlation is unlikely for steric and other reasons, the location of the gold colloids in Fig. 6 visualizes the distribution of the grafting sites on the CNT surface. Grafting occurs along the whole length of the nanotubes. The grafting sites are probably associated with the presence of graphene edges and defect sites in the CNT sidewalls. However, transmission electron microscopy studies (not shown) failed to demonstrate a conclusive relationship between the gold nanoparticles locations and defect structures, probably due to the difficulty of imaging all types of defects in the relevant orientation. Fig. 6(a) also implies that functionalisation occurs in a relatively homogeneous fashion. On the macroscale, this observation is supported by the relatively good repeatability of the TGA experiments (typically ±10% relative error in grafting ratios). However, for industrial scale-up, issues associated with heat and organic reactant transport will need to be addressed. Various other functional vinyl compounds were grafted onto commercial CNTs.
 | ||
| Fig. 6 SEM images of in-house CNTs grafted with (a) MTEMA and (b) LMA after exposure to a dispersion of gold nanoparticles, followed by thorough washing in both cases. | ||
Compared to both the untreated parent material and the ‘air-exposed’ control sample, the solvent dispersibility of the modified CNTs improved significantly, in a range of solvents (Table 1; the particular examples cover a broad spectrum of solvent polarity), depending on the functionality of the covalently-attached moiety. For instance, the introduction of methyl methacrylate (MMA) oligomers led to significantly increased dispersibility in butyl acetate but reduced dispersion in more polar ethanol. On the other hand, grafting of the CNTs with 2-(dimethylamino)ethyl methacrylate (DMAEMA) resulted in poor dispersion in butyl acetate but markedly improved dispersibility in acidic aqueous solution due the electrostatic stabilisation of CNTs by protonated amine groups. This change in dispersibilty is consistent with the altered thermodynamic surface properties of the grafted CNTs, as measured by IGC. The dispersive surface energy of the grafted CNTs roughly correlates with the grafting ratio (Table 1); with increasing coverage of the highly energetic graphitic surface, γd decreases. Changes in the KD/KA ratios after grafting (Table 1) indicate altered surface characters due to the introduction of new functional surface groups. For instance, relative to the parent material, the KD/KA ratio decreased for CNTs modified with acrylic acid (AA), indicating a more electron accepting character, but increased for the DMAEMA-grafted CNTs, implying a more electron donating surface. These findings demonstrate that our methodology is an efficient and flexible route to tailor the CNT surface chemistry, rendering our approach a useful tool to adapt as-produced CNTs to potential applications, such as sensor networks, filters, electrochemical device electrodes, or catalysis support materials. CNT functionalisation is required to improve the interfacial compatibility of the nanotubes with other components (such as electrolytes, reaction solvents, or composite matrices)10,42,43 or to provide a direct function (for example, analyte binding, catalytic activity, photocharge generation).44–46 As an illustration, the gold-tagged CNTs, presented in Fig. 6, could be useful supported (electro)catalyst materials in their own right.
Our proposed thermochemical modification treatment offers several advantages over conventional CNT grafting strategies. Firstly, our approach can exploit existing surface oxide defect sites that are characteristic of most (commercial) CVD-grown MWCNTs, minimising the framework damage and associated degradation of intrinsic properties. Conventional defect chemistry, in contrast, tends to use a strong preliminary acid oxidation that is known to cut and etch nanotubes.13 Many other literature grafting and functionalisation reactions attack intact CNT sidewalls and, thereby, directly increase the defect concentration.47 Secondly, the grafting reaction was found to be highly efficient, typically recovering at least 90% of the original CNTs, in grafted form. The observed material loss can be attributed to handling issues at the current experimental scale, and can be expected to decrease significantly at larger scales. Although only a small fraction of the monomer reservoir is reacted per cycle, the overall monomer utilisation is high. The grafting efficiency of our methodology in the liquid setup was determined to be at least 99% for the MMA-grafted commercial CNTs; i.e. less than 1% of the original monomer was lost due to formation of free homopolymer, enabling the re-use of monomer for future grafting cycles. The high grafting efficiency can be attributed to initiation and propagation of the grafting reaction through surface-bound radical intermediates. In principle, the treatment can be carried out without creating any chemical waste; depending on the application, excess monomer may either remain in the final product or be removed through evaporation under vacuum, making time-consuming filtration and washing procedures redundant. Thirdly, and perhaps most importantly, our methodology can be carried out in a pure gas-phase reaction when comparatively volatile monomers, such as MMA and acrylonitrile (AN), are used under vacuum conditions. The grafting ratios, as determined by TGA, and surface properties, as determined by gas chromatography, are comparable to the corresponding products obtained using the liquid-phase setup (Table 1). This particular approach has the additional advantage that the inhibitor does not have to be removed from the monomer reservoir. Consequently, unreacted monomer remains stabilized against self-polymerisation. The gas-phase set-up is also easily scalable and compatible with many existing CNT synthesis processes, rendering our methodology more economically viable than existing grafting approaches.
Conclusions
The thermochemical approach introduced in this edge article is a generic method to modify CNTs whilst minimising damage to the graphitic framework. The key step of the treatment is the activation of CNTs at high temperatures under inert atmosphere or in a vacuum, which results in the desorption of oxygen-containing functional groups at pre-existing defect sites and the generation of surface radicals. The reactive sites can be utilised to initiate radical polymerisation of functional monomers leading to the grafting of oligomers. The grafting ratios and average oligomer chain lengths of the modified CNTs lie between 1–8 wt% and 1–70 monomeric units, respectively; the results depend on the reactivity and flexibility of the organic reactant selected. Both dispersion studies and IGC characterisation indicate significantly altered CNT surface properties depending on the grafting ratio and functionality of the monomer. Our functionalisation approach combines versatility with the technological advantages of solvent-free reactions and scalability; the treatment is of high efficiency due to the “grafting-from” reaction mechanism. The functionalisation can also be carried out as a pure gas-phase reaction, rendering it potentially compatible with existing gas-phase processes for the commercial production of CNTs. Significantly, the same approach should also apply to other technologically important carbons, including blacks and fibers.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) and Defence Science and Technology Laboratory (DSTL). We would also like to thank Richard Winchester for acquiring the TEM images.Notes and references
- M. C. Ma, T. C. Brown and B. S. Haynes, Surf. Sci., 1993, 297, 312 CrossRef CAS.
- A. Frumkin, Kolloid-Z., 1930, 51, 123 CrossRef CAS.
- Y. A. Zarifyanz, V. F. Kiselev, N. N. Lezhnev and O. V. Nikitina, Carbon, 1967, 5, 127 CrossRef.
- P. M. Ajayan, Chem. Rev., 1999, 99, 1787 CrossRef CAS.
- B. Njuguna, Adv. Eng. Mater., 2003, 5, 769 CrossRef.
- J. N. Coleman, U. Khan and Y. K. Gunko, Adv. Mater., 2006, 18, 689 CrossRef CAS.
- Y. J. Lu, J. Li, J. Han, H. T. Ng, C. Binder, C. Partridge and M. Meyyappan, Chem. Phys. Lett., 2004, 391, 344 CrossRef CAS.
- B. Yoon and C. M. Wai, J. Am. Chem. Soc., 2005, 127, 17174 CrossRef CAS.
- E. Katz, ChemPhysChem, 2004, 5, 1084 CrossRef CAS.
- C. A. Dyke and J. M. Tour, J. Phys. Chem. A, 2004, 108, 11151 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- C. N. R. Rao and A. Govindaraj, Nanotubes and Nanowires, ed. P. O'Brian, H. Kroto, and H. Craighead, Royal Society of Chemistry, Cambridge, 2005 Search PubMed.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS.
- M. Hamon, H. Hui, P. Bhowmik, M. E. Itkis and R. C. Haddon, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 333 CrossRef CAS.
- M. Holzinger, O. Vostrowsky, A. Hirsch, F. Hennrich, M. Kappes, R. Weiss and F. Jellen, Angew. Chem., Int. Ed., 2001, 40, 4002 CrossRef CAS.
- Y. S. Chen, R. C. Haddon, S. Fang, A. M. Rao, P. C. Eklund, W. H. Lee, E. C. Dickey, E. A. Grulke and R. E. Smalley, J. Mater. Res., 1998, 13, 2423 CAS.
- J. Bahr, J. Yang, D. V. Kosynkin, M. J. Bronikowski, R. E. Smalley and J. M. Tour, J. Am. Chem. Soc., 2001, 123, 6536 CrossRef CAS.
- S. Campidelli, C. Sooambar, E. Lozano Diz, C. Ehli, D. M. Guldi and M. Prato, J. Am. Chem. Soc., 2006, 128, 12544 CrossRef CAS.
- C. M. Homenick, G. Lawson and A. Adronov, Polym. Rev., 2007, 47, 265 Search PubMed.
- X. D. Lou, C. Detrembleur, V. Sciannamea, C. Pagnoulle and R. Jerome, Polymer, 2004, 45, 6097 CrossRef CAS.
- Y. Q. Liu, Z. L. Yao and A. Adronov, Macromolecules, 2005, 38, 1172 CrossRef CAS.
- C. E. Hamilton, J. R. Lomeda, Z. Z. Sun, J. M. Tour and A. R. Barron, Nano Res., 2010, 3, 138 Search PubMed.
- Z. L. Yao, N. Braidy, G. A. Botton and A. Adronov, J. Am. Chem. Soc., 2003, 125, 16015 CrossRef CAS.
- M. Dehonor, K. Masenelli-Varlot, A. Gonzalez-Montiel, C. Gauthier, J. Y. Cavaille and M. Terrones, J. Nanosci. Nanotechnol., 2007, 7, 3450 CrossRef CAS.
- J. Cui, W. P. Wang, Y. Z. You, C. H. Liu and P. H. Wang, Polymer, 2004, 45, 8717 CrossRef CAS.
- T. Nakajima, S. Kasamatsu and Y. Matsuo, Eur. J. Solid State Inorg. Chem., 1996, 33, 831 CAS.
- M. Q. Tran, C. Tridech, A. Alfrey, A. Bismarck and M. S. P. Shaffer, Carbon, 2007, 45, 2341 CrossRef CAS.
- Z. Y. Hou, B. C. Cai, H. Liu and D. Xu, Carbon, 2008, 46, 405 CrossRef CAS.
- A. Bismarck, C. Wuertz and J. Springer, Carbon, 1999, 37, 1019 CrossRef CAS.
- R. Andrews, D. Jacques, A. M. Rao, F. Derbyshire, D. Qian, X. Fan, E. C. Dickey and J. Chen, Chem. Phys. Lett., 1999, 303, 467 CrossRef CAS.
- R. Menzel, A. Lee, A. Bismarck and M. S. P. Shaffer, Langmuir, 2009, 25, 8340 CrossRef CAS.
- M. Voll and H. P. Boehm, Carbon, 1970, 8, 741 CrossRef CAS.
- J. Carlsson, F. Hanke, S. Linic and M. Scheffler, Phys. Rev. Lett., 2009, 102, 166104 CrossRef.
- S. Hamdi, B. Hamdi, Z. Kessaissia, H. Barthel, H. Balard and J. B. Donnet, J. Chromatogr., A, 2002, 969, 143 CrossRef CAS.
- M. Kosaka, T. W. Ebbesen, H. Hiura and K. Tanigaki, Chem. Phys. Lett., 1994, 225, 161 CrossRef CAS.
- F. Beuneu, C. l'Huillier, J. P. Salvetat, J. M. Bonard and L. Forro, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 5945 CrossRef CAS.
- C. Goze-Bac, S. Latil, P. Lauginie, V. Jourdain, J. Conard, L. Duclaux, A. Rubio and P. Bernier, Carbon, 2002, 40, 1825 CrossRef CAS.
- B. Corzilius, K. P. Dinse, K. Hata, M. Haluska, V. Skakalova and S. Roth, Phys. Status Solidi B, 2008, 245, 2251 CrossRef CAS.
- K. Sendt and B. S. Haynes, J. Phys. Chem. C, 2007, 111, 5465 CrossRef CAS.
- T. J. Frankcombe and S. C. Smith, Carbon, 2004, 42, 2921 CrossRef CAS.
- K. S. Coleman, S. R. Bailey, S. Fogden and M. L. H. Green, J. Am. Chem. Soc., 2003, 125, 8722 CrossRef CAS.
- W. Zhang, J. K. Sprafke, M. L. Ma, E. Y. Tsui, S. A. Sydlik, G. C. Rutledge and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 8446 CrossRef CAS.
- L. Q. Liu, A. H. Barber, S. Nuriel and H. D. Wagner, Adv. Funct. Mater., 2005, 15, 975 CrossRef CAS.
- R. M. Lucente-Schultz, V. C. Moore, A. D. Leonard, B. K. Price, D. V. Kosynkin, M. Lu, R. Partha, J. L. Conyers and J. M. Tour, J. Am. Chem. Soc., 2009, 131, 3934 CrossRef CAS.
- P. Singh, J. Kumar, F. M. Toma, J. Raya, M. Prato, B. Fabre, S. Verma and A. Bianco, J. Am. Chem. Soc., 2009, 131, 13555 CrossRef CAS.
- S. M. Kaniber, M. Brandstetter, F. C. Simmel, I. Carmeli and A. W. Holleitner, J. Am. Chem. Soc., 2010, 132, 2872 CrossRef CAS.
- X. M. Sui, S. Giordani, M. Prato and H. D. Wagner, Appl. Phys. Lett., 2009, 95, 233113 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Names and acronyms of the reactants used for thermochemical grafting, additional information on how the grafting ratio, Δ, was determined, the derivatives of the TGA profiles presented in Fig. 5, EPR spectra of heat-treated and air-exposed CNTs at room temperature, and additional characterisation data for the grafted CNTs presented in Table 1 (numerical values for the KA and KD numbers, for the IG/ID ratios and for the concentrations of dispersed CNTs in various, additional solvents). See DOI: 10.1039/c0sc00287a |
| This journal is © The Royal Society of Chemistry 2010 |