A tangled web—interconnecting pathways to amorphadiene and the amorphene sesquiterpenes†
Young J.
Hong
and
Dean J.
Tantillo
*
University of California–Davis, Davis, CA 95616, USA. E-mail: tantillo@chem.ucdavis.edu; Fax: +1 530-752-8147; Tel: +1 530-754-5635
First published on 13th August 2010
Abstract
Quantum chemical studies on the formation of amorphadiene and the amorphene sesquiterpenes are described. These natural products are commonly thought to arise from distinct pathways involving initial 1,6- and 1,10-cyclization of farnesyl diphosphate, respectively. We have found, using density functional calculations, that the pathway usually invoked for amorphadiene formation is not only energetically feasible, but is also the energetically favored pathway to the amorphenes as a result of a low energy (two-step) 1,5-hydride transfer involving an unusual carbocation containing a 3-center 2-electron [C⋯H⋯C]+ bonding array.
Introduction
The complex, polycyclic hydrocarbon cores of terpene/terpenoid natural products are assembled in the active sites of terpene synthase enzymes using exceedingly simple (acyclic, achiral, repetitive) precursors.1 Many biochemical2–5 and theoretical6–11 studies have provided insights into the mechanisms of these amazing transformations, but many questions (both general and system-dependent) still linger. Herein we describe a theoretical study on the formation of the isomeric amorphadiene and amorphene sesquiterpenes (1–4), showing that previously proposed pathways to these two groups of terpene natural products (blue and red in Scheme 1; in this report, we focus only on diastereomers that lead to 1–4),2,3,5,12,13 which diverge from each other at their beginning, actually converge just before their conclusion.14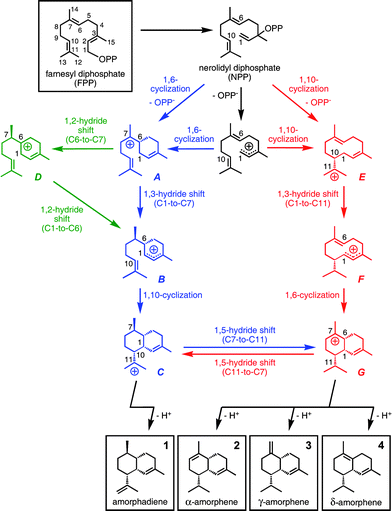 | ||
| Scheme 1 | ||
Methods
All calculations were performed with GAUSSIAN03.15 All geometries were optimized using the B3LYP/6-31+G(d,p) method.16 All stationary points were characterized by frequency calculations and reported energies include zero-point energy corrections (unscaled). Intrinsic reaction coordinate (IRC) calculations were used for further characterization of all transition state structures.17 The B3LYP method is known to perform reasonably well in the prediction of the geometries and behavior of carbocations.6–11,18–20 We also include mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) energies for comparison, since B3LYP tends to underestimate the relative stabilities of cyclic structures versus acyclic isomers.11 These energies include unscaled zero-point energy corrections from B3LYP/6-31+G(d,p) frequency calculations. We have used these methods previously to study other terpene-forming carbocation rearrangement reactions (and have compared them to the results of other methods).7–9,14,18–20 All energies for intermediate structures and transition state structures in this report are expressed in kcal mol−1 and are relative to that of the bisabolyl cation conformer A0 (Fig. 1), whose energy is set to [0.00] kcal mol−1. Structure A0 is used here as a reference so that the energies described herein can be compared directly to those of the carbocations encountered en route to other bisabolyl cation-derived sesquiterpenes examined previously.7h,8,9 Structural drawings were produced using Ball & Stick.21 Atom numbering indicated in the structures in this report refers to that of FPP (Scheme 1). Throughout the manuscript, bold numbers are used to label natural products and bold capital letters are used to label carbocations. Numbers following letters for carbocations are used to label different conformations.![Structure of (R)-bisabolyl cation conformer A0, whose energy is set to [0.00] kcal mol−1 for all energy comparisons.](/image/article/2010/SC/c0sc00333f/c0sc00333f-f1.gif) | ||
| Fig. 1 Structure of (R)-bisabolyl cation conformer A0, whose energy is set to [0.00] kcal mol−1 for all energy comparisons. | ||
Results and discussion
The most commonly proposed mechanism for amorphadiene formation (Scheme 1, blue)2,3 involves the conversion of farnesyl diphosphate (FPP) to nerolidyl diphosphate (NPP), which is then converted, upon loss of pyrophosphate and 1,6-cyclization, into the bisabolyl cation (A).22 A 1,3-hydride shift (or two consecutive 1,2-hydride shifts via cation D, the homobisabolyl cation;2,3,13,23Scheme 1, green) leads to cation B,24 which can then cyclize to form cation C, the immediate precursor to amorphadiene (1). Note that in this mechanism, only tertiary and allylic cations would be formed and π-bonds are traded for σ-bonds in the steps in which allylic conjugation is lost.A pathway involving initial 1,10-cyclization (Scheme 1, red) has been proposed as a possible alternative to the pathway to amorphadiene involving initial 1,6-cyclization,3,25 but is more commonly invoked as a route to the amorphenes.5,12,13 In this pathway, loss of pyrophosphate and 1,10-cyclization of nerolidyl diphosphate lead to the (Z,E)-germacradienyl cation (E),26 which then undergoes a 1,3-hydride shift (or two consecutive 1,2-hydride shifts)2,3,5 to produce cation F,27 which can then undergo a transannular attack of the C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C7 π-bond onto C1 to produce cation G,28 the immediate precursor to the amorphenes (2–4). Again, only tertiary and allylic cations would be formed in this mechanism.
C7 π-bond onto C1 to produce cation G,28 the immediate precursor to the amorphenes (2–4). Again, only tertiary and allylic cations would be formed in this mechanism.
Interconversion of cations C and Gvia a net 1,5-hydride shift would allow either of the blue and red pathways to produce the amorphenes and/or amorphadiene.29
Initial 1,6-cyclization
A complete pathway (Scheme 1, blue) from bisabolyl cation conformer A1 to one conformer of cation C based on our calculations is shown in Fig. 2.30 The first step in this pathway, 1,3-hydride transfer from C1 to C7 (A1→B1), is predicted to have a low barrier, consistent with previous work on such reactions.31 Note that the hydride shift occurs via the “interior” of the acyclic chain of bisabolyl cation conformer A1, which results in the R configuration of the resulting stereogenic center in cation B1 if one starts with (R)-A1.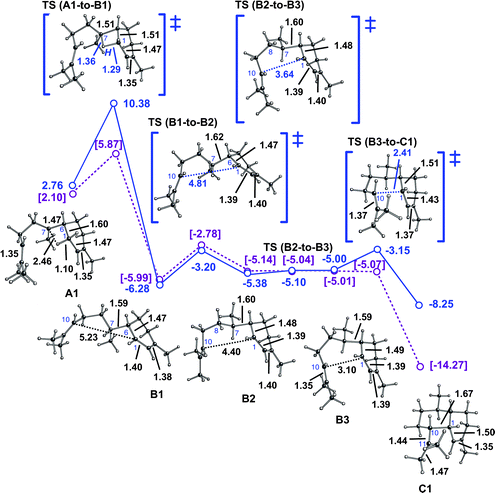 | ||
| Fig. 2 Conversion of A1 to C1via a pathway involving a 1,3-hydride shift. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in blue and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). Note that the B3-to-C1 transition state structure is lower in energy than B3 at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level (mPW1PW91 single point energies on geometries optimized with B3LYP), suggesting that this conversion may actually be barrierless. | ||
The next bond-making step is 1,10-cyclization, but cation B1 must first undergo several low-barrier conformational changes (B1→B2→B3; Fig. 2) so that C1 and C10 are appropriately oriented for cyclization,32i.e., are in close proximity to each other. Attack by the C10C11 π-bond on the pro-R face of the cationic center at C1 leads to C1via a chair-like transition state structure with the C(CH3)2 group at an equatorial position. Like most previously described transition state structures for cation-alkene cyclization reactions (reactions that are consistently exothermic, since they involve trading a π-bond for a σ-bond), this transition state structure is quite early.6b–c,9,11,33 The cyclization is essentially barrierless once the appropriate conformation of cation B is formed.14b
Overall, the A1-to-C1 rearrangement is exothermic by more than 10 kcal mol−1 and is not associated with a significant barrier. Thus, it seems that this mechanism for amorphadiene formation is indeed feasible as generally formulated unless the enzyme active site in which the cations are generated is shaped so as to preclude the formation of productive conformations.34
A cationic two-step
As mentioned above, the direct 1,3-hydride shift could be avoided by a detour involving two sequential 1,2-hydride shifts (Scheme 1, green). A computed pathway (Scheme 1, green and blue) from one bisabolyl cation conformer (A2) to one conformer of cation Cvia cation D is shown in Fig. 3. The first hydride shift, to form cation D1, is predicted to have a barrier of ∼6–7 kcal mol−1. Note that this hydride shift over the “exterior” face of the acyclic chain of the bisabolyl cation leads directly to a conformation that is productive for the subsequent 1,2-hydride shift. The transition state structure for the second 1,2-hydride shift is similar in energy to that of the first and it leads directly to a conformer of cation B that is productive for 1,10-cyclization (which is again found to be essentially barrierless).14b Note that in the conformer of cation C formed by this route, the C1–C10 σ-bond is significantly elongated (to 1.77 Å), likely as a result of through-bond coupling between the cationic center at C11 and the C2C3 π-bond (and perhaps also to alleviate a steric clash between hydrogens on C5 and C9, which are only 2.04 Å apart in C2).14c,35–37
 | ||
| Fig. 3 Conversion of A2 to C2via a pathway involving two 1,2-hydride shifts. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in green and blue and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). Note that the B4-to-C2 transition state structure is lower in energy than B4 at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level (mPW1PW91 single point energies on geometries optimized with B3LYP), suggesting that this conversion may actually be barrierless. | ||
The overall barrier for this two-step process is similar to that for the one-step process described above. Consequently, we suspect that an enzyme that chooses between them likely does so via conformational preorganization. In fact, the two-step process has been ruled out for amorphadiene formation in Artemisia annua L.,3 since FPP labeled with a deuterium at C1 produces amorphadiene with a deuterium at C7. Note, though, that this does not rule out a pathway involving initial 1,10-cyclization (vide infra) or two-step pathways for amorphadiene formation in other organisms.2,3,13
Initial 1,10-cyclization
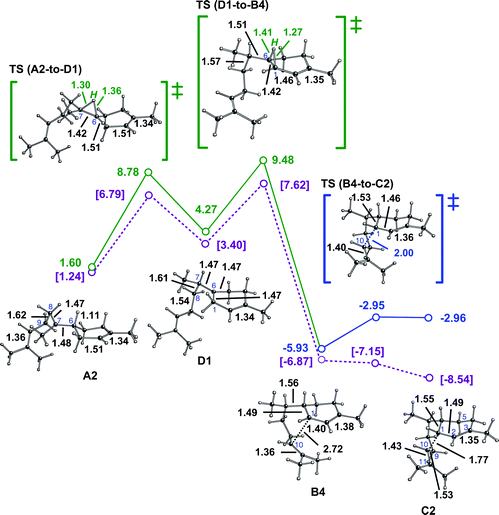
A computed reaction pathway to cation G from a productive conformer of the (Z,E)-germacradienyl cation (E1) is shown in Fig. 4.30 Although the barrier for the 1,3-hydride transfer is again small,31 the absolute energy of its transition state structure is considerably higher than that encountered in the pathway via the bisabolyl cation (vide supra). This is likely the result of strain in this transition state structure, and the germacradienyl cation preceding it, expected based on the fact that they contain medium-sized rings with several sp2 centers.38 Hydride transfer leads to a structure, cation F1, with allylic conjugation, however, which counterbalances some of this strain. Similar to the cation-alkene cyclization steps described above, the F1-to-G1 reaction is essentially barrierless (note the presence of a short cation-π contact and associated long C6C7 π-bond in F1, which also contributes to its relatively low energy), is exothermic, and produces a structure with significant through-bond coupling.14c,35,36 Overall, this pathway is again significantly exothermic and has only a small overall barrier, assuming that the (Z,E)-germacradienyl cation can actually be formed in the enzyme active site.39
 | ||
| Fig. 4 Conversion of E1 to G1. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in red and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). Note that the F1-to-G1 transition state structure is lower in energy than F1 at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level (mPW1PW91 single point energies on geometries optimized with B3LYP), suggesting that this conversion may actually be barrierless. | ||
Bridging the gap
The pathways involving initial 1,6- and 1,10-cyclization lead to different products—amorphadiene and the amorphenes, respectively—but if cations C and G could interconvert, then each pathway could lead to amorphadiene and/or the amorphenes. This would be important, since the highest energy transition state structure on the 1,6-cyclization pathway is considerably lower in energy than that on the 1,10-cyclization pathway.We located a transition state structure that looked like an early transition state structure for a 1,5-hydride transfer between C11 and C7 of cation C (Fig. 5; note that the productive conformer for the hydride shift differs from that shown in Fig. 2,34 but is similar to it in energy and can be formed from it via conformational changes involving low barriers; see ESI† for details). IRC calculations indicated, however, that this transition state structure actually connects cation C3 to a cation, H1, that contains a delocalized 3-center 2-electron [C⋯H⋯C]+ bonding array. Despite resembling a transition state structure for the desired 1,5-hydride shift, cation H1 is actually a minimum. Other cations with acyclic [C⋯H⋯C]+ bonding arrays have been detected in NMR experiments and examined theoretically,40 but, to our knowledge, this is the first such cation found for a biologically relevant carbon skeleton, although arguably not the most unusual.7c,14c,19,20 Although the C3-to-H1 conversion is endothermic, it is only somewhat so and involves only a small barrier.
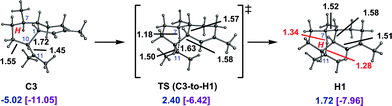 | ||
| Fig. 5 Conversion of C3 to H1. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in blue and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). | ||
A similar cation, H2, was located when looking for transition state structures for 1,5-hydride shifts starting from conformers of cation G (Fig. 6). Again only a small barrier need be overcome to form the delocalized cation.41
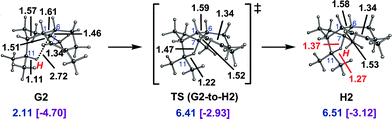 | ||
| Fig. 6 Conversion of G2 to H2. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in blue and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). | ||
Cations H1 and H2 are conformers of each other and their interconversion (via the transition state structure shown in Fig. 7) involves a small barrier. Thus, the pathways involving initial 1,6- and 1,10-cyclization are, indeed, entangled. In that cations H1 and H2 and the transition state structures that connect them to each other and to C3 and G2 are all considerably lower in energy than the highest energy transition state structure encountered on the 1,10-cyclization pathway, it is clear that pathways to amorphadiene and the amorphenes that involve initial 1,6-cyclization are more accessible energetically than those that involve 1,10-cyclization, at least in the absence of enzymatic intervention.
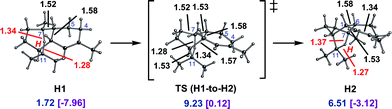 | ||
| Fig. 7 Interconversion of H1 and H2. Computed distances (Å) and energies (kcal mol−1) are shown (B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in blue and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in plum and brackets). | ||
Implications
To our knowledge, the co-production of amorphadiene and an amorphene by a single enzyme has not yet been reported. Nonetheless, on the basis of our results, we suggest that perhaps the minor products produced in small amounts by the enzymes that produce either amorphadiene or the amorphenes should be reexamined. Moreover, our results suggest a mechanism by which an amorphadiene synthase could evolve into an amorphene synthase or vice versa.Acknowledgements
We gratefully acknowledge UC Davis, the National Science Foundation's CAREER program, and the National Science Foundation's Partnership for Advanced Computational Infrastructure (Pittsburgh Supercomputer Center) for support and Dr Michael Lodewyk for helpful comments.Notes and references
- (a) D. W. Christianson, Chem. Rev., 2006, 106, 3412–3442 CrossRef CAS; (b) D. W. Christianson, Curr. Opin. Chem. Biol., 2008, 12, 141–150 CrossRef CAS; (c) R. Croteau, Chem. Rev., 1987, 87, 929–954 CrossRef CAS; (d) D. E. Cane, in Sesquiterpene Biosynthesis: Cyclization Mechanisms, Elsevier, 1999, vol. 2, pp. 155–200 Search PubMed; (e) D. E. Cane, Chem. Rev., 1990, 90, 1089–1103 CrossRef CAS; (f) D. E. Cane, Acc. Chem. Res., 1985, 18, 220–226 CrossRef CAS; (g) E. M. Davis and R. Croteau, Top. Curr. Chem., 2000, 209, 53–95 CAS.
- S. Picaud, P. Mercke, X. He, O. Sterner, M. Brodelius, D. E. Cane and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 448, 150–155 CrossRef CAS.
- S.-H. Kim, K. Heo, Y.-J. Chang, S.-H. Park, S.-K. Rhee and S.-U. Kim, J. Nat. Prod., 2006, 69, 758–762 CrossRef CAS.
- E.g.: (a) D. E. Cane and T. E. Bowser, Bioorg. Med. Chem. Lett., 1999, 9, 1127–1132 CrossRef CAS; (b) M. M. Ravn, R. M. Coates, R. Jetter and R. B. Croteau, Chem. Commun., 1998, 21–22 RSC; (c) M. M. Ravn, R. M. Coates, J. E. Flory, R. J. Peters and R. Croteau, Org. Lett., 2000, 2, 573–576 CrossRef CAS; (d) D. C. Williams, B. J. Carroll, Q. Jin, C. D. Rithner, S. R. Lenger, H. G. Floss, R. M. Coates, R. M. Williams and R. Croteau, Chem. Biol., 2000, 7, 969–77 CrossRef CAS; (e) R. B. Croteau, C. J. Wheeler, D. E. Cane, R. Ebert and H. J. Ha, Biochemistry, 1987, 26, 5383–9 CrossRef CAS; (f) D. E. Cane, J. L. Pawlak and R. M. Horak, Biochemistry, 1990, 29, 5476–90 CrossRef CAS; (g) D. E. Cane, P. C. Prabhakaran, J. S. Oliver and D. B. McIlwaine, J. Am. Chem. Soc., 1990, 112, 3209–3210 CrossRef CAS; (h) N. Bülow and W. A. König, Phytochemistry, 2000, 55, 141–168 CrossRef CAS.
- D. Arigoni, Pure Appl. Chem., 1975, 41, 219–245 CrossRef.
- (a) C. Jenson and W. L. Jorgensen, J. Am. Chem. Soc., 1997, 119, 10846–10854 CrossRef CAS; (b) B. A. Hess, Org. Lett., 2003, 5, 165–167 CrossRef; (c) B. A. Hess and L. Smentek, Org. Lett., 2004, 6, 1717–1720 CrossRef CAS; (d) M. Weitman and D. T. Major, J. Am. Chem. Soc., 2010, 132, 6349–6360 CrossRef CAS; (e) H. Oikawa, K. Nakamura, H. Toshima, T. Toyomasu and T. Sassa, J. Am. Chem. Soc., 2002, 124, 9145–9153 CrossRef CAS; (f) R. K. Allemann, N. J. Young, S. Ma, D. G. Truhlar and J. Gao, J. Am. Chem. Soc., 2007, 129, 13008–13013 CrossRef CAS; (g) A. F. Barrero, M. M. Herrador, J.-L. Lopez-Perez, J. F. Arteaga and J. Catalan, Org. Lett., 2009, 11, 4782–4785 CrossRef CAS; (h) J. A. Faraldos, P. E. O'Maille, N. Dellas, J. P. Noel, R. M. Coates, J. Am. Chem. Soc., 132, 4281–4289 Search PubMed; (i) J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377–392 CrossRef CAS.
- (a) M. W. Lodewyk, P. G. Gutta and D. J. Tantillo, J. Org. Chem., 2008, 73, 6570–6579 CrossRef CAS; (b) G. A. Ho, D. H. Nouri and D. J. Tantillo, J. Org. Chem., 2005, 70, 5139–5143 CrossRef CAS; (c) P. Gutta and D. J. Tantillo, J. Am. Chem. Soc., 2006, 128, 6172–6179 CrossRef CAS; (d) P. Gutta and D. J. Tantillo, Org. Lett., 2007, 9, 1069–1071 CrossRef CAS; (e) S. C. Wang and D. J. Tantillo, Org. Lett., 2008, 10, 4827–4830 CrossRef CAS; (f) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 5375–5386 CrossRef CAS; (g) Y. J. Hong and D. J. Tantillo, Nat. Chem., 2009, 1, 384–389 Search PubMed; (h) Y. J. Hong and D. J. Tantillo, Org. Biomol. Chem., 2009, 7, 4101–4109 RSC; (i) Y. J. Hong, D. J. Tantillo, Org. Biomol. Chem. 10.1039/c0ob00167h.
- Y. J. Hong and D. J. Tantillo, Org. Lett., 2006, 8, 4601–4604 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2009, 131, 7999–8015 CrossRef CAS.
- J. B. A. Hess, J. Am. Chem. Soc., 2002, 124, 10286–10287 CrossRef.
- S. P. T. Matsuda, W. K. Wilson and Q. Xiong, Org. Biomol. Chem., 2006, 4, 530–543 RSC.
- (a) C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078–2089 CrossRef CAS; (b) D. B. Little and R. B. Croteau, Arch. Biochem. Biophys., 2002, 402, 120–135 CrossRef CAS.
- F. Lopez-Gallego, S. A. Agger, D. Abate-Pella, M. D. Distefano and C. Schmidt-Dannert, ChemBioChem, 2010, 11, 1093–1106 CrossRef CAS.
- (a) This is Part 7 in our series of reports on “Theoretical Studies on Farnesyl Cation Cyclization.” For parts 1–6, see ref. 7a, 7c, 7e, 7h, 8 and 9. For reviews with leading references to theoretical work by us and others on closely related systems, see: (b) D. J. Tantillo, J. Phys. Org. Chem., 2008, 21, 561–570 CrossRef CAS and (c) D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847–2854 RSC.
- M. J. Frisch, et al., GAUSSIAN 03 (Revision D.01), Gaussian, Inc.. Pittsburgh, PA, 2003 Search PubMed (full reference in ESI†).
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, J. Org. Chem., 2007, 72, 8877–8881 CrossRef CAS.
- M. D. Bojin and D. J. Tantillo, J. Phys. Chem. A, 2006, 110, 4810–4816 CrossRef CAS.
- P. Gutta and D. J. Tantillo, Angew. Chem., Int. Ed., 2005, 44, 2719–2723 CrossRef CAS.
- N. Müller, A. Falk and G. Gsaller, Ball & Stick v. 4.0a12, Molecular Graphics Application for MacOS Computers, Johannes Kepler University, Linz, 2004 Search PubMed.
- A vast array of sesquiterpenes can be derived from the bisabolyl cation. For our previous theoretical work on pathways to some of these, see ref. 7h, 8 and 9.
- Cation D may also be the immediate precursor to β- and γ-curcumene: ( (a) C. G. Jones, E. L. Ghisalberti, J. A. Plummer and E. L. Barbour, Phytochemistry, 2006, 67, 2463–2468 CrossRef CAS; (b) T. G. Köllner, P. E. O'Maille, N. Gatto, W. Boland, J. D. Gershenzon and J. Degenhardt, Arch. Biochem. Biophys., 2006, 448, 83–92 CrossRef; (c) T. G. Köllner, C. Schnee, J. Gershenzon and J. Degenhardt, Plant Cell, 2004, 16, 1115–1131 CrossRef ) and, via further rearrangements, to a variety of other sesquiterpenes9.
- Cation B may also be the immediate precursor to β-sesquiphellandrene, zingeberene and γ-curcumene.23a–c β-Sesquiphellandrene and zingeberene have been detected amongst the mixture of products from the amorphadiene synthase cloned from Artemissia annua L2,3.
- W. Parker, J. S. Roberts and R. Ramage, Q. Rev. Chem. Soc., 1967, 21, 331–363 RSC.
- Cation E may also be the immediate precursor to germacrene A and B12.
- Cation F may also be the immediate precursor to germacrene C and D4h,12.
- We have previously examined other reactions of cation G that lead to the ylangenes, sativene and cyclosativene7a.
- A related 1,4-hydride shift is frequently proposed to occur in trichodiene biosynthesis,1a,d–f but quantum chemical calculations suggest that this process is associated with a substantial barrier and is unlikely to occur7h,8.
- As in previous cases, the acyclic C1–C2–C3 allylic cation derived via loss of pyrophosphate from NPP (Scheme 1) was not found as a minimum.8,9 This is consistent with a direct displacement reaction,4b–c but our calculations do not rule out formation of the allylic cation in an enzyme active site.
- See, for example, our theoretical study on presilphiperfolanol formation and references therein7e.
- Although such conformational changes are often not necessary in terpene-forming reactions, i.e., the conformer produced in one step is productive for the next step, from time to time they are9.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- Similar pathways involving different conformers of cations A, B and C were also examined. See ESI† for details.
- R. Hoffmann, A. Imamura and W. J. Hehre, J. Am. Chem. Soc., 1968, 90, 1499–1509 CrossRef CAS.
- R. Hoffmann, Acc. Chem. Res., 1971, 4, 1–9 CrossRef CAS.
- Deprotonation of C2 to form amorphadiene was modeled using ammonia. See ESI† for details.
- For leading references, see: (a) V. Prelog, Pure Appl. Chem., 1963, 6, 545–560 CrossRef CAS; (b) J. F. Liebman and A. Greenberg, Chem. Rev., 1976, 76, 311–365 CrossRef; (c) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS; (d) M. A. Winnik, Chem. Rev., 1981, 81, 491–524 CrossRef CAS; (e) W. C. Still and I. Galynker, Tetrahedron, 1981, 37, 3981–3996 CrossRef CAS.
- Similar pathways involving other conformers (including isopropyl group rotamers) were also examined. See ESI† for details.
- For leading references, see: (a) R. Ponec, G. Yuzhakov and D. J. Tantillo, J. Org. Chem., 2004, 69, 2992–2996 CrossRef CAS; (b) J. E. McMurry and T. Lectka, Acc. Chem. Res., 1992, 25, 47–53 CrossRef CAS.
- Deprotonation of cations H1 and H2 using an ammonia model was also examined. Complexation of these cations via C–H⋯X interactions involving certain hydrogens did prompt their geometries to become more localized. See ESI† for details. Related geometric changes upon complexation have been described previously7a,f,i,9,18,19.
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates and energies of computed structures, IRC plots, information on additional calculations referred to in references and complete ref. 15. See DOI: 10.1039/c0sc00333f |
| This journal is © The Royal Society of Chemistry 2010 |