X-ray snapshots for metalloporphyrin axial ligation†
Lin X.
Chen
*ab,
Xiaoyi
Zhang
c,
Erik C.
Wasinger
a,
Jenny V.
Lockard
a,
Andrew B.
Stickrath
a,
Michael W.
Mara
b,
Klaus
Attenkofer
c,
Guy
Jennings
c,
Grigory
Smolentsev
d and
Alexander
Soldatov
d
aChemical Sciences and Engineering Division, 9700 South Cass Avenue, Argonne, Illinois 60439, USA. E-mail: lchen@anl.gov; Fax: + 1 6302529389; Tel: + 1 6302523533
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
cX-ray Science Division, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
dPhysics Department, Southern Federal University, Rostov-na-Donu, 344090, Russia
First published on 30th September 2010
Abstract
Axial ligation mechanisms of a metalloporphyrin, nickel(II) tetramesitylporphyrin (NiTMP), were investigated by static and transient X-ray absorption spectroscopy at Ni K-edge (8.333 keV). A surprisingly broad (i.e. ∼1.4 eV) linewidth for the 1s → 3dx2-y2 transition in the ground state was attributed to strong geometry dependent 3d molecular orbital (MO) energies due to coexisting conformers in solution. The broad distribution of 3d MO energy levels enables transient degeneracy of the 3dz2 and 3dx2-y2 MOs to produce a temporary vacancy in the 3dz2 MO which favors axial ligation. Photoexcitation also induces the vacancy in the 3dz2 MO, leading to a more than two-fold enhancement in the axial ligated species. Therefore, a unified axial ligation mechanism for both the ground and excited state is proposed based on the elucidation of the excited state structural dynamics, which will have a broad impact in understanding and controlling axial ligation in enzymatic reactions and molecular catalysis involving transient axial ligation.
Introduction
Metalloporphyrins have versatile light activated functions including light harvesting, photoinduced electron and energy transfer, and photocatalysis.1–3 Their photocatalytic activities are often based on light triggered electronic configuration changes that enable the axial ligation and de-ligation with substrates, from or to which electrons can be transferred. Therefore, revealing the transient electronic and nuclear structural identities during photoexcitation triggered ligation/de-ligation processes is crucial for understanding structural influences on catalytic functions and control of photocatalytic reactions through structural factors.The metalloporphyrin chosen in our study was Ni(II) tetramesitylporphyrin (NiTMP). Molecular dynamics and structures of nickel(II) porphyrins were extensively studied by ultrafast transient absorption spectroscopy,4–11 resonance Raman spectroscopy,12–20 X-ray transient absorption spectroscopy21–25 and quantum mechanical calculations.25–29 Together, these studies showed severe non-planar distortions of the porphyrin macrocycle due to a mismatch of the smaller Ni(II) ion with the larger porphyrin macrocycle to which the Ni(II) was coordinated. Several optical signatures were attributed to the non-planar distortion and conformational heterogeneity.16,18,20,30,31 Meanwhile, Holten and others have found that the axial ligation of Ni(II) porphyrin can be induced by photoexcitation in a solvent mixture of ligating pyridine (or piperidine) and non-ligating toluene.32,33 The proposed reaction pathway involves a π → π* transition, followed by intramolecular energy transfer coupled with intersystem crossing to the T1 state. Deactivation of the T1 state proceeds via two routes back to the ground state: a) dual axial ligation followed by ligand release and return to the ground state, and b) direct return to the ground state (Fig. 1a, and 1b).4,5,33 The same group later measured photoinduced axial ligation of nickel dodecaarylporphyrins (NiDPP) in several ligating solvents.32 They proposed that NiDPP formed a penta- or hexa-coordinate intermediate which then underwent a dual-decay process to either a relaxed six-coordinate intermediate or the ground state.32 In spite of this plausible proposal, the transient optical absorption study itself could neither resolve the structures involved in the axial ligation process nor distinguish single or dual ligated species.
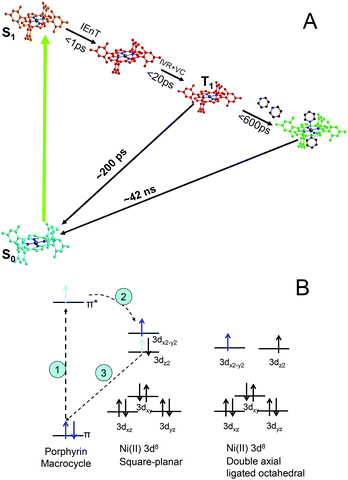 | ||
| Fig. 1 (A) The excited state axial ligation pathways for NiTMP in the presence of ligating pyridine; and (B) time sequence for the electronic configuration changes: 1) photoexcitation induces π → π* transition, 2) intramolecular energy transfer, and 3) 3dz2 electron fills the hole in the ground state π MO. | ||
The intramolecular energy transfer results in the electron movement from the π* molecular orbital (MO) to the unoccupied antibonding 3dx2-y2 dominated MO with concurrent electron movement from the occupied 3dz2 to the vacated π MO. This scheme was verified by Zamyatin et al. through two-photon excitation transient absorption measurements where the first photon induced a π → π* transition and the second photon, delayed until after the simultaneous intramolecular energy transfer and reoccupation of the π MO, induced the π → π* transition to prove the recovery of the hole in the π MO.34 The resulting electronic configuration of singly occupied 3dz2 and 3dx2-y2 was identified in our previous studies with the X-ray transient absorption (XTA) method.23
In spite of these previous studies, the excited state Ni(II) porphyrin ligation structure and time sequence as well as mechanisms for the excited and ground state ligation are not completely clear. Correlations between the electronic state transition and the metal center ligation state need to be further explored in order to rationally design systems with desirable catalytic properties. Results from this study will provide insight into excited state structural control for metal ligation/deligation involved in photocatalytic processes.
Using both X-ray transient absorption spectroscopy (XTA)35–40 and optical transient absorption spectroscopy (OTA), we previously studied electronic and nuclear structures of the T1 excited state of NiTMP without the axial ligation in neat non-ligating toluene.11,23 We observed for the first time the direct evidence of singly occupied 3dz2 and 3dx2-y2 MOs in the T1 excited state, and determined the energy difference between 1s →3dz2 or 3dx2-y2 transitions, as well as the energy shifts of 3dx2-y2 and 4pz MOs in the T1 state from that of the S0 state. Meanwhile, the Ni–N bond elongation in the T1 state was also resolved, suggesting an increased ionic radius for Ni(II) in the excited state and a more planar macrocycle conformation.
In this report, we focus on the molecular structural dynamics of the NiTMP excited state in a new process, the photoinduced axial ligation of NiTMP* (* denotes the photoexcited state) with pyridine in a mixed toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pyridine solvent. Both electronic and nuclear configurations have been monitored, and the results not only shed light on the time sequence of excited state structural changes, but also reveal a unified mechanism for Ni(II) porphyrin ligation in the T1 and S0 state. The correlations between the structural diversity and the excited state properties resulting from this study are important for elucidation and control of photocatalytic reactions such as fuel generation by sunlight in particular.
:pyridine solvent. Both electronic and nuclear configurations have been monitored, and the results not only shed light on the time sequence of excited state structural changes, but also reveal a unified mechanism for Ni(II) porphyrin ligation in the T1 and S0 state. The correlations between the structural diversity and the excited state properties resulting from this study are important for elucidation and control of photocatalytic reactions such as fuel generation by sunlight in particular.
Experimental methods
1. Samples
NiTMP was purchased from Frontier Scientific without further purification. The solvents, toluene and pyridine, were obtained from a solvent dispenser under anaerobic atmosphere freshly collected before the experiments.2. Experimental setup and conditions of XTA measurements
XTA (or LITR-XAS laser-initiated time-resolved XAS) measurements were carried out at Beamline 11ID-D, the Advanced Photon Source (APS) of Argonne National Laboratory.22,23,35–37,40 The details of the experimental setup have been described in the Supplemental Information of our previous publication.37 Briefly, the laser pump, X-ray probe cycle was at 1 kHz repetition rate with 0.6 mJ/pulse at 527 nm and a FWHM of 5 ps at the sample. The laser excitation energy coincides perfectly with the Q-band of NiTMP, inducing the S0 → S1 transition. The X-ray probe pulses were extracted from a train of electron bunches circulating in the storage ring at 272 kHz under a hybrid timing mode. An eight-element solid-state germanium array detector (Canberra) was used to collect X-ray fluorescence signals with Soller slits and a cobalt filter combination to minimize elastic scattering signals. The detector array outputs were amplified and converted through single channel analyzers, and then were split into three portions connecting to three sets of scalers, respectively. The first portion was ungated signals from all the X-ray pulses, which was used for extracting the ground state spectrum, the second portion was signals electronically gated with the laser pulse at 1 kHz for the laser-on spectrum, and the third portion was similarly gated to the second portion except that the gate was set at 147 μs after the laser pulse for the laser-off ground state spectrum. The second and the third portions of the signals were used to generate the difference spectrum because the two have the same signal to noise ratio and the same counting statistics. The delay time between the laser and the X-ray pulses was adjusted by a programmable delay line (PDL-100A-20NS, Colby Instruments) based on a fast Si PIN diode detector signal positioned at the sample location to detect both X-ray and laser pulses.3. UV-vis spectroscopy and solvent titration
The UV-vis spectra of NiTMP in different solvents were measured by a Shimatzu UV-1601 spectrophotometer with a 2 mm path cuvette at room temperature. Because the axial ligation in the ground state NiTMP in a mixed solvent with toluene and pyridine shifts both the Soret- and the Q-band to distinctly different wavelengths from the non-ligated species, the concentration ratio for the ligated and unligated species can be determined. Concentration ratios of unligated to ligated NiTMP in the solvent mixtures were determined by multiple peak fits of the UV-vis spectra obtained from a titration process where the volume ratio of pyridine:toluene was altered.
4. Transient optical spectroscopy of NiTMP in the photoinduced ligation process
The OTA measurements have been carried out using three different setups to cover the time scales from 100 fs to 100 ns as well as to mimic specific sample environments. The femtosecond TA spectra were measured with a Ti:sapphire laser system with a regenerative amplifier and an optical parametric amplifier (OPA) as described elsewhere.11 The optical pump pulse was the output of the OPA adjusted to 527 nm. The probe white light generated by focusing a few μJ of Ti:sapphire amplifier output onto a sapphire disk was split into two beams for the probe and probe reference, respectively. The FWHM of the instrument response function was 120 fs. The excited state population and kinetics on the picosecond time scale were obtained by the single wavelength picosecond pump–probe transient absorption spectroscopy with the sample conditions (beam size, concentration, and laser pulse energy, etc.) very close to that for the XTA experiments. The 527 nm laser was split into two beams: 5% was used for the probe while the rest was used for the pump. The transient absorption measurements on nanosecond time scale were measured using the same pump source as used in the XTA experiments, while the probe was a white light source from a xenon flash lamp.
5. Density functional theory (DFT) calculations
Energies of molecular orbitals (MOs) were calculated using the Amsterdam Density Functional program suite (ADF2007.01)41,42 in the framework of density functional theory (DFT). Within a non-relativistic approach the electronic configuration of the molecule was described by an uncontracted triple-ζ basis set of Slater-type orbitals with one polarization function. Energies were calculated using Perdew-Wang exchange correlation potential43,44 within the generalized gradient approximation. Displacements of atoms were performed along normal coordinates corresponding to low frequency vibrational modes of the porphyrin macrocycle corresponding to A1g, B1u and B2u symmetry representations. Calculations of vibrational modes were published previously.45Results
1. Steady-state and transient optical absorption measurements for the NiTMP ligation
The unligated and ligated NiTMP have very different Soret- and Q-band positions (Fig. 2). The structures of Ni(II) porphyrins in the presence of ligating solvents, such as pyridine and piperidine, has long been debated. It is uncertain whether the ligated species exist in a single axial ligated square-pyramidal or a double axial ligated six-coordinate structure. The unligated NiTMP has a single Q-band absorption peak at 527 nm, whereas the ligated NiTMP has two peaks at 563 and 600 nm, respectively. Therefore, the 527 nm laser pulses only excite the unligated ground state NiTMP and hardly perturb the ligated ground state NiTMP. Meanwhile, the ligation also changes the Soret-band absorption from a broader peak at 414 nm for the unligated NiTMP to a narrower peak at 434 nm for the ligated NiTMP. Multiple peak curve fitting of the Soret bands in the UV-vis spectrum of NiTMP in neat pyridine was used to determine the fraction of the ligated species. The UV-vis spectra of NiTMP in toluene as a function of the pyridine concentration during the titration suggested that the fraction of ligated species in the ground state NiTMP is ∼16% in a 3:1 v/v pyridine/toluene mixture and ∼51% in neat pyridine.
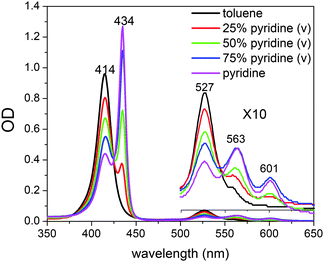 | ||
| Fig. 2 UV-vis spectra of NiTMP in mixed solvents (toluene:pyridine) at room temperature. | ||
Because of these distinct spectral features, we were able to determine the concentration change of unligated NiTMP as a function of the pyridine concentration, and hence deduce the equation for the chemical equilibrium of the axial ligation.
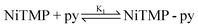
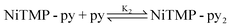
The detailed processes of extracting K1 and K2 are included in the ESI†. Using the values of the equilibrium constants, K1 = 0.043M−1, and K2 = 0.10M−1, the ligation is most likely a two-step process, as shown above and as seen in previous studies of tetra-meso-substituted nickel porphyrins.46 Although the optical absorption features between ligated and unligated NiTMP are distinctively different, those between monoligated, NiTMP-py and dual ligated NiTMP-py2 are not easily distinguishable. Based on the two-step ligation mechanism and the values of K1 and K2, the concentrations of NiTMP, NiTMP-py and NiTMP-py2 as functions of the pyridine concentration, [py], can be obtained (see the ESI†), which gives a total of 16% of ligated species, NiTMP-py and NiTMP-py2. Although there have been discussions in the literature about the single axial ligated species, we are not able to distinguish NiTMP-py and NiTMP-py2 at a volume ratio of toluene:pyridine = 3:1 by ground state X-ray absorption spectroscopy (XAS), although the calculated concentrations for NiTMP-py at this pyridine concentration is higher then that for NiTMP-py2. However, we did not observe the expected pre-edge enhancement of the 1s → 3d transition in the XANES spectra due to the Ni 3d-4p mixing which is significantly stronger in a single axial ligated square-pyramidal geometry.47 This may be due to the overall low concentration of ligated species relative to the unligated species.
The single wavelength picosecond pump–probe transient absorption measurements at 527 nm revealed that the ground state recovery kinetics can be described by multiple exponential functions with three time constants, τ1 = 20 ps, τ2 = 255 ps, and τ3 > 10 ns. We attribute τ1 to the intramolecular vibrational relaxation and vibrational cooling based on previous studies.9,11 According to the measured ground state recovery kinetics, the branching ratio for the excited state population to a long-lived presumably ligated intermediate or to the ground state is 24% to 76%, respectively. Hence, the time constants of decaying to the ground state and to a long-lived intermediate are about 330 ps and 1 ns, respectively. The long-lived presumably ligated intermediate decays to the unligated ground state with a time constant τ3 = 42 ns as determined by the nanosecond transient absorption measurements.
2. The pre-edge linewidth analysis at Ni K-edge
The pre-edge features of Ni K-edge, which appear as weak peaks below the transition edge energy, have been assigned as the dipole-forbidden and quadrupole-allowed 1s → 3d transitions.48 Previous studies of the angular dependence of the 1s → 3d peak intensities in a CuCl4 (D4h) single crystal as a function of the orientation of the sample with respect to the polarization direction of the X-rays revealed the correlations of the transitions with different 3d orbitals in the molecular frame.47,49 Although the intensities of pure 1s → 3d transitions due to the quadrupole coupling are much weaker than the dipole allowed transitions (∼1:100), they can gain intensity when the metal coordination loses centrosymmetry allowing for Ni 3d-4p mixing. Hence, these pre-edge features can be used to reveal the 3d MO energy levels and the non-centrosymmetric coordination geometry.47
The pre-edge region of the XANES spectra at the Ni K-edge is presented in Fig. 3. Because Ni(II) (3d8) is in a porphyrin macrocycle and coordinated with four N atoms on the xy-plane, its 3d MOs have the energy distribution with an empty 3dx2-y2 at the highest energy and the other four fully occupied 3dz2, 3dxy, and doubly degenerate 3dxz and 3dyz in descending energies (Fig. 1). Therefore, only one pre-edge peak corresponding to 1s → 3dx2-y2 transition was observed in the ground state S0 spectrum. As revealed by our previous study,23 the pre-edge spectrum for the Ni(II) center in the T1 state has two peaks corresponding respectively to the transitions 1s → 3dx2-y2, and 1s → 3dz2. This was the first direct XAS evidence showing the two singly occupied MOs, 3dx2-y2 and 3dz2 at the T1 state. The energy difference between the two transitions appeared to be ∼2.2 eV without deconvolution of the intrinsic energy resolution of the beamline. The peak positions are shifted slightly relative to each other when comparing the spectra taken at the X-ray probe pulse delay time of t = 0 and 100 ps. Because the current time resolution of the experiment is limited by the X-ray pulse duration of 160 ps, the absolute delay time between the pump and the probe is not accurate. However, the relative delay can be controlled precisely to 0.5 ps.
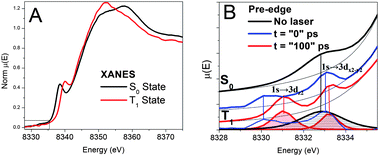 | ||
| Fig. 3 A) XANES spectra and B) pre-edge spectra of NiTMP in toluene at different states obtained from XTA measurements. The bottom of B) displays the linewidth comparison for the 1s to 3d transitions for the S0 (shaded black) and T1 (shaded red) states, and vertical lines connect the pre-edge features before and after the removal of the background absorption shown by the thin black lines. | ||
Noticeable differences in the bandwidth for the pre-edge peaks for the S0 and T1 states, which have been recently confirmed by new measurements at a different beamline, Beamline 7IDC of the APS, turn out to be very important in understanding the axial ligation mechanism. The pre-edge peak corresponding to the 1s → 3dx2-y2 transition in the S0 state has an apparent linewidth >2eV (FWHM), whereas the two pre-edge peaks corresponding to the 1s → 3dx2-y2, and 1s → 3dz2 transitions in the T1 state have linewidths of 1.0–1.2 eV. The comparison of the linewidths between the two spectra verifies that the S0 1s → 3d pre-edge peak width is larger than the energy resolution of the beamline instrumentation, and reflects the intrinsic properties of NiTMP in solution at room temperature. Obviously, we cannot rule out that the 1s → 3dx2-y2, and 1s → 3dz2 transitions in the T1 state may have even narrower linewidths, and the observed 1.0–1.2 eV linewidth (FWHM) may be limited by the energy resolution of the beamline instrumentation. The origin of the linewidth difference and its implications will be discussed later.
3. Transient XANES spectra of NiTMP during the photoinduced ligation process
Fig. 4 displays the transient XANES spectra of 1.5 mM NiTMP in a mixed solvent of toluene and pyridine (volume ratio = 3:1) during the photoinduced ligation at different delay times, focusing on the 1s → 4pz transition at 8337.5 eV. This peak feature has proven to be very sensitive to the ligation states of metalloporphyrins.22,25,28 In a previous study on the photodissociation of two axial piperidine ligands from NiTPP (TPP = tetraphenylporphyrin),22 the almost completely ligated hexa-coordinated Ni(II) has no detectable intensity at 8337.5 eV whereas the square-planar NiTPP has a very sharp feature due to the 1s → 4pz transition.
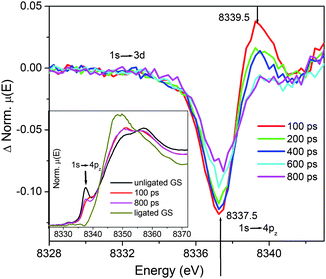 | ||
| Fig. 4 Transient XANES spectra of NiTMP in a mixed solvent (toluene:pyridine = 3:1 by volume ratio). The edge feature focused on 1s → 4pz for monitoring the spectral evolution as a function of the pump–probe delay times. Inset shows the XANES spectra measured at 100 ps, 800 ps, and the XANES spectra of pure ligated and unligated NiTMP in the ground state. | ||
According to the optical absorption spectra of NiTMP in the solvent mixture of toluene:pyridine (v:v) = 3:1 or [py] = 3.11 M (Fig. 2), ∼16% NiTMP became NiTMP-py or NiTMP-py2, and the fraction of NiTMP-py + NiTMP-py2 became ∼51% in neat pyridine. Based on the optical and the XANES spectra of the ground state NiTMP in the mixed toluene:pyridine (3:1 by volume), neat toluene and neat pyridine, the XANES spectra for the 100% dual-ligated NiTMP-py2 can be extracted (Fig. 4 inset). The XANES spectrum of NiTMP-py2 (Fig. 4 inset) has no distinct 1s → 4pz transition peak because the corresponding transition has a higher energy which shifts the peak feature to an energy as high as the whiteline transition. XANES spectra measured at nominally 100 ps and 800 ps delays after the laser pump pulse are also depicted in Fig. 4 inset and will be discussed later. In the XTA spectrum taken at a 100 ps delay, a bleaching occurs at 8337.5 eV due to the reduction of the 1s → 4pz transition from a reduced population for the unligated ground state upon the photoexcitation. A new absorption appears at 8339.5 eV, which is at the exact energy as the 1s → 4pz transition of the unligated excited NiTMP from the XTA measurement on NiTMP in neat toluene (Fig. 3).23 During 200–400 ps after the excitation, both the excited state absorption and the ground state bleaching decreases. By the delay time of 600–800 ps after the excitation, the ground state bleaching is still changing while the absorption at 8339.5 eV remains largely unchanged.
4. 3d MO energies as a function of the NiTMP conformation: DFT calculations
The apparent ∼2 eV linewidth of the pre-edge peak assigned to the 1s → 3dx2-y2 transition observed in the ground state NiTMP (Fig. 3) suggests a possibility of a broader energy distribution of unoccupied orbitals than previously anticipated. Several comprehensive studies have addressed the conformational diversity of Ni(II) porphyrins in solution at room temperature by optical absorption and resonance Raman spectroscopies as well as quantum mechanical calculations.10,12,17,30 While the Q-band absorption of various Ni(II) porphyrins are shown to be sensitive to the conformation and the ligation status of the Ni(II) center, there has not been a direct way to measure the distribution of the 3d MO energy as a function of molecular conformations.In order to investigate whether the 1s → 3d transition energies are sensitive to the NiTMP conformations, a series of DFT calculations were carried out for a group of NiTMP conformations with non-planar distortions, such as saddled, twisted, and domed, with Ni–N distances adjusted accordingly. The distortions made in the calculations were generated through the displacement of normal modes of the molecules and were within the range of those shown in the literature.24 However, it is beyond the scope of this report to calculate accurate values for the 1s → 3dz2 or 3dx2-y2 transition energies using DFT methods. The trend of the 3d MO energy dependence on the conformation can be shown and the energies of 3dz2 and 3dx2-y2 MOs for distorted NiTMPs are listed in Table I and illustrated in Fig. 5 to demonstrate qualitatively the energy variation.
| 3dz2 energy/eV | 3dx2-y2 energy/eV | ΔE3d/eV | 1s energy/eV | ΔE1s-3dx2-y2/eV | description | |
|---|---|---|---|---|---|---|
| 1 | −4.648 | −3.186 | 1.462 | −8119.95 | 8116.74 | Planar, rNi–N 1.98 Å |
| 2 | −4.651 | −2.937 | 1.714 | −8118.90 | 8115.96 | Planar, rNi–N 1.95 Å |
| 3 | −4.659 | −2.677 | 1.919 | −8119.96 | 8117.29 | Planar, rNi–N 1.91 Å |
| 4 | −5.212 | −4.039 | 1.173 | −8120.59 | 8116.55 | Saddling rNi–N 1.94 Å |
| 5 | −4.734 | −2.792 | 1.942 | −8120.07 | 8117.28 | Saddling, rNi–N 1.91 Å |
| 6 | −4.562 | −2.362 | 2.200 | −8118.42 | 8116.06 | Ruffling, rNi–N 1.91 Å |
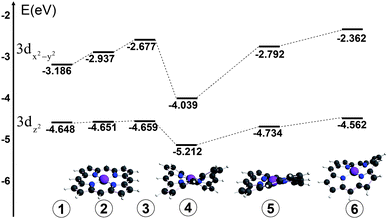 | ||
| Fig. 5 Energy diagram of 3dx2-y2 and 3dz2 MOs for different conformers of NiTMP that may exist at room temperature, by DFT calculations. | ||
Discussion
1. Coordination geometry dependent 3d MO energies and its implications
The apparent ∼2 eV linewidth for the 1s → 3dx2-y2 transition in the ground state NiTMP in non-coordinating toluene and its contrast with approximately 1.0–1.2 eV linewidths for the 1s → 3dx2-y2, 3dz2 transitions in the T1 state confirms that the former has a linewidth exceeding the intrinsic energy resolution of the experiment and a broad energy distribution of the unoccupied 3dx2-y2 MO. Two questions immediately emerge, 1) what is the actual energy distribution in the 1s → 3dx2-y2 transition of the S0 state, and 2) what causes such a seemingly wide energy distribution?In order to resolve the actual energy distribution range for the 1s → 3dx2-y2 transition in the S0 state of NiTMP, two assumptions are made: a) the excited state pre-edge peaks that have much narrower linewidths represent the intrinsic energy resolution limited linewidth convoluted with the lifetime broadening of the transitions, and b) the probabilities for interconvertable conformations of NiTMP at 298 K are nearly equal due to their small activation barriers of kT ∼ 0.03 eV. Under these assumptions, the broad 1s → 3dx2-y2 peak in the S0 state with apparent ∼2.2 eV FWHM can be decomposed into multiple Lorentzian peaks with the same area and width due to the 1s → 3dx2-y2 transitions from different conformers. The minimal number of identical peaks required to fit the experimental data is three, with a maximum separation of ∼1.4 eV between the peaks, suggesting a maximum energy separation of ∼1.4 eV between the lowest and the highest energy transitions (Fig. 6).
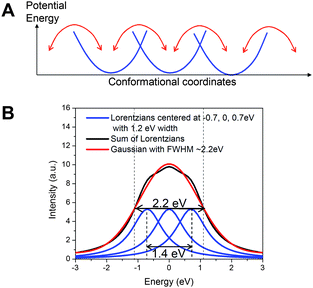 | ||
| Fig. 6 A) The isoenergetic multiple conformation model of the ground state NiTMP in solution at room temperature; B) The model fit to a Gaussian peak mimicking the experimentally observed 1s → 3dx2-y2 transition feature at the pre-edge region of the Ni K-edge with a FWHM of ∼2.2 eV. The fitting indicates that at least three Lorentzian peaks with 1.2 eV width (the assumed intrinsic energy resolution limit obtained from the T1 state pre-edge features in Fig. 3) are needed. | ||
Next we look for the origins of this unexpectedly large energy difference in the 1s → 3dx2-y2 transition energy. The 1.4 eV transition energy spread is ∼ 50 times higher than the energy barrier of kT at 298 K, ∼0.03 eV or 208 cm−1. The question is how different conformers of NiTMP interconvert in solution at room temperature with only ∼0.03 eV energy barriers can afford the energy spread of ∼1.4 eV in energies of the 1s → 3dx2-y2 transitions. In order to answer this question, we carried out a series of DFT calculations on selected NiTMP conformations distorted from the planar pseudo D4h symmetry (Table I). Because the average Ni–N bond distance will be longer in the planar conformation than those of domed, twisted, and ruffled non-planar conformations, the bond distances alter with the non-planar distortions. According to the calculated results, the energy spread for the 3dx2-y2 MO can be as large as ∼1.1 eV, and the energy gap spread between the 1s and 3dx2-y2 MOs can be as large as ∼1.3 eV, similar to experimental observation. A complete simulation of the energy distribution needs sampling of all possible molecular structures with a correct population distribution, which is beyond the scope of this report. Nevertheless, even a few representative structures and energies (Fig. 5) can help to rationalize the experimental observation. Clearly, the differentiation in the linewidth for the 1s → 3d transition in the S0 and T1 states suggests a much broader conformation distribution in the former than the latter. This result agrees with the Ni–N bond distance changes from the S0 to T1 state obtained from our previous study, where the average Ni–N distance in the T1 state increases by 0.08 Å. The severe non-planar distortion is a result of the shorter Ni–N distanced due to the mismatch between the Ni(II) radius and the size of the porphyrin macrocycle cavity. The interconversion between different severely distorted non-planar conformations inevitably samples a broader or more diverse range of structures, resulting in a wider potential well in the S0 state. In contrast, the longer Ni–N distances in the T1 state imply a better match between the Ni(II)* radius and the porphyrin cavity, resulting in a more planar conformation, sampling a much narrower range of coordinates in a narrower potential well. This analysis is consistent with our experimental observations.
In addition, we measured the XANES spectrum of the S0 state at 15 K (not shown) when the potential barriers kT for conformation interconversion would be ∼0.5–0.6 eV, 20 times of that at room temperature. The pre-edge peak corresponding to the 1s → 3dx2-y2 transition narrows by ∼0.4 eV without splitting, and shifts to ∼0.5 eV higher in peak energy. According to Fig. 5, the conformations with higher 3dx2-y2 MO energies are those with more planar conformations and longer Ni–N distances, assuming the 1s MO energy changes due to the conformation change can be neglected as an approximation. This agrees qualitatively with the observation that a planar Ni(II)TMP conformation in the T1 state has a higher energy in the 1s → 3dx2-y2 transition. The pre-edge results provide direct evidence for the MO energy variation as a function of the conformations, as well as the potential well widths for the ground S0 and the excited T1 states, which were unknown before. The conformation dependent energy variation of the 3d MO energies revealed by this study has a direct impact on our understanding of the ground and excited state axial ligation as described below.
2. Photoinduced axial ligation with coordinating solvent molecules
The excited state ligation process was monitored by the amplitude of the 1s → 4pz transition peak. The photoexcitation initially creates the singlet S1 state of NiTMP which undergoes intramolecular energy transfer to an electronic configuration of singly occupied 3dz2 and 3dx2-y2 MOs as shown in our previous experiments.23 One important question is what the time sequence is for the photoinduced ligation of NiTMP in the mixed solvent. In other words, does the ligation take place as the S1 state is generated or after the triplet state is produced through the intersystem crossing? Or does the ligation happen before, during or after vibration relaxation?The dual ligated NiTMP has no 1s → 4pz feature at the K-edge because the transition has been shifted to a higher energy due to the elevation of the 4pz MO energy.22,49 If NiTMP forms penta- or hexa-coordinate species after photoexcitation, we should see a decrease in the sharp 1s → 4pz peak at 8337.5 eV. However, the 1s → 4pz transition peak intensity of NiTMP in the mixed solvent did not decrease at 100 ps after the photoexcitation, but shifted to a higher energy (inset of Fig. 4). The XTA spectrum at 100 ps delay (Fig. 4) is very similar to that for the T1 state of NiTMP in neat toluene without the ligation.23 Therefore, we conclude that solvent ligation happens after the formation of the T1 excited state and the new absorption peak at higher energy is due to the formation of excited unligated NiTMP. Between 100 ps and 400 ps, the transient absorption intensity at 8339.5 eV reduced and almost goes to zero after 600 ps, which indicates the diminishing population of the unligated excited state after 600 ps. This agrees with the optical TA measurements that lifetime of T1 excited state (unligated) is about 255 ps in the mixed solvent. During the 600–800 ps time delay from excitation, the 1s → 4pz peak at 8337.5 eV reappears with a reduced intensity. The time evolution of the 1s → 4pz peak is an indication of the branched pathways for the T1 state in the presence of pyridine ligating molecules as mentioned earlier. Because the dual ligated NiTMP has shifted its 1s → 4pz peak to a higher energy under the whiteline transition, it will not contribute to the intensity at 8337.5 eV. Therefore, the reduced intensity at 8337.5 eV indicates that the ligation has occurred, and can be used to extract the remaining unligated population. The ground state bleaching of the unligated 1s → 4pz transition at 8337.5 eV could last for several tens of nanoseconds because the photoinduced ligated NiTMP decays to the ground state with a time constant of 42 ns.
These results provide answers to the previous questions regarding the time sequence of the photoinduced ligation. The solvent ligation happens with a time constant of ∼1 ns, long after the vibrational relaxation of the T1 state. This time constant agrees with a solvent diffusion process. Because the ligation process is diffusion limited, its intrinsic diffusion rate determines the branching ratio of the T1 state after its creation. The relatively long-lived ligated species also indicate that the ligated species is a triplet state that requires tens of nanoseconds to release the ligands and return to the S0 state. So far, we have not seen solid evidence of penta-coordinated NiTMP-py in any of the transient XANES spectra when pyridine is present. The pre-edge data have not shown any enhanced 1s → 3d transition intensity expected for a square-pyramidal species.
Ni(II) porphyrins are known to undergo double axial ligation in the S0 state in the presence of ligating molecules. Meanwhile, Ni(II) porphyrins have been shown to undergo wavelength dependent photoinduced ligation/deligation.32,33 It has been proposed by Holten and coworkers that the doubly ligated ground state NiTPP has degenerate singly occupied 3dx2-y2 and 3dz2 MOs where the lone pair electrons from nitrogenated ligating molecules will donate electrons to the Ni(II) center from above and below the porphyrin plane to achieve the dual ligation. Then the questions are 1) what is the driving force for the empty 3dx2-y2 and the occupied 3dz2 orbitals of the starting Ni(II) porphyrin to convert to degenerate singly occupied MOs to allow axial ligation, and 2) what is the time sequence for the ligation with respect to the electronic configuration change? These questions are important for our understanding of the correlations between electronic and nuclear structures of the molecule, as well as controlling molecular reactivity through structural variations, which is an important attribute in catalysis.
The axial ligation of Ni(II) porphyrins is related to their conformations in terms of steric hindrance imposed by the side groups in the meso- and β-positions, as well as out-of-plane distortion. It is intuitive that ligation is increasingly difficult as the macrocycle becomes more non-planar or the side groups bulkier, inhibiting access of the ligating molecule to the Ni(II) center. The energy spread revealed by the ground state XANES spectrum provides direct evidence of the energy spread of 1s → 3dx2-y2 transitions as a function of conformation. The 3dx2-y2 energy fluctuation with the conformation as well as the coexistence of several possible conformations suggest a possibility of transiently degenerate 3dx2-y2 and 3dz2 MOs, which would allow one electron hopping from the fully occupied 3dz2 to 3dx2-y2. When the resulting electron vacancy in the 3dz2 MO encounters the lone pair electrons from the nitrogenated ligand molecule, it accommodates the ligation along the z-axis and forms a relatively stable doubly ligated species. Therefore, the dynamic fluctuation revealed by the XANES spectrum becomes the driving force for the ground state ligation with pyridine.
In the solvent mixture used in the XTA measurements, there was ∼16% NiTMP-py + NiTMP-py2 and ∼84% NiTMP before the laser excitation. The laser pulse at 527 nm initially excited ∼70% of NiTMP, yielding ∼59% (0.84 × 0.70 = 0.59) excited NiTMP (Fig. 7). From the difference in the XTA spectrum at 800 ps after the excitation (Fig. 4), the reduced intensity at 8337.5 eV corresponds to an additional 14% or more of total NiTMP molecules were converted to NiTMP-py + NiTMP-py2 yielding a total of 30% ligated species (Fig. 7). Apparently, the photoexcited state favors solvent ligation compared to the ground state. This enhancement due to the photoexcitation can be explained by our previous results on the pre-edge 1s → 3d transitions for the T1 state where the 3dz2 MO becomes singly occupied. The vacated 3dz2 MO can accommodate the axial ligation more easily, and the double ligation can take place, resulting in triplet degenerate 3dz2 and 3dx2-y2 as expected for the pseudo-Oh symmetry in the hexa-coordinated coordinated Ni(II).
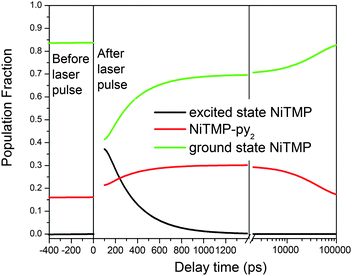 | ||
| Fig. 7 The constructed population fractions of different species of NiTMP before and after the laser excitation as a function of the delay time based on the optical and X-ray transient absorption spectroscopic results. The delay time axis has both linear and log scales. | ||
From the picosecond TA measurements, the decay time constant of the unligated T1 state in the presence of pyridine was determined to be 330 ps, which is significantly longer than that of ∼200 ps in neat toluene. Such difference in the T1 state lifetime is indicative of solvation of NiTMP pyridine that has a different polarity than toluene. Although nearly 60% of the T1 state in mixed solvent decays to the ground state without the axial ligation, the presence of pyridine may solvate NiTMP differently than toluene causing the change in the excited state lifetime. Such subtle structural change due to the solvation may also explain the absence of the dual pre-edge peaks in the mixed solvent.
Conclusions
The X-ray absorption spectroscopic studies on NiTMP in solution revealed a strong dependence of the 3d MO energies with the interconvertable conformations at room temperature in solution, resulting in an energy spread for 1s → 3dx2-y2 transitions up to ∼1.4 eV. The 3d MO energy fluctuations due to the interconversions among the conformers produce transient degeneracies between the two upper 3d MOs, a filled 3dz2 and an empty 3dx2-y2. Consequently, the transient degeneracy enables one of the 3dz2 electrons hopping onto the empty 3dx2-y2, resulting in two singly occupied MOs in favor of axial ligation in the ground state.X-ray transient absorption spectroscopic studies on the photoexcited NiTMP in a mixed solvent of non-ligating toluene and ligating pyridine revealed the mechanism for the photoinduced ligation. The photoexcitation of the Q-band created vibrational hot singly occupied 3dz2 and 3dx2-y2 MOs that are also in favor of axial ligation. However, axial ligation with the excited state NiTMP does not take place immediately but is a diffusion limited process with a time constant of ∼1 ns in the particular solvent mixture used in our study. The photoexcitation shifted the ligation equilibrium NiTMP* + 2py ↔ NiTMP(py)2* to the right with a two-fold enhancement in the population of the ligated species compared to that in the ground state. Therefore, the photoinduced axial ligation was also promoted by a vacancy in the 3dz2 along the direction of axial bonding.
This study provides a unified mechanism for axial ligation driven by the vacancy in the 3dz2 MO in both the ground and excited state NiTMP which may be applicable to other metalloporphyrins and is expected to have a general impact in designing photocatalysts for promoting binding to the metal center in a well defined orientation in order to achieve desirable products.
Acknowledgements
This work is supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U. S. Department of Energy under contracts DE-AC02-06CH11357 (LXC, ECW, JVL, ABS, MWM). The DFT calculation part is supported by a joint grant to AS and LXC from the U.S. Civilian Research and Development Fund (RUC1-2870-RO-07). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357 (XZ, AK, GJ).Notes and references
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, 2002 Search PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS.
- J. Rosenthal, J. Bachman, J. L. Dempsey, A. J. Esswein, T. G. Gray, J. M. Hodgkiss, D. R. Manke, T. D. Luckett, B. J. Pistorio, A. S. Veige and D. G. Nocera, Coord. Chem. Rev., 2005, 249, 1316–1326 CrossRef CAS.
- D. Kim and D. Holten, Chem. Phys. Lett., 1983, 98, 584–589 CrossRef CAS.
- D. Kim, C. Kirmaier and D. Holten, Chem. Phys., 1983, 75, 305–322 CrossRef CAS.
- E. W. Findsen, K. Alston, J. A. Shelnutt and M. R. Ondrias, J. Am. Chem. Soc., 1986, 108, 4009–4017 CrossRef CAS.
- M. Hoshino, Inorg. Chem., 1986, 25, 2476–2478 CrossRef CAS.
- E. W. Findsen, J. A. Shelnutt and M. R. Ondrias, J. Phys. Chem., 1988, 92, 307–314 CrossRef CAS.
- H. S. Eom, S. C. Jeoung, D. Kim, J.-H. Ha and Y.-R. Kim, J. Phys. Chem. A, 1997, 101, 3661–3669 CrossRef CAS.
- C. M. Drain, S. Gentemann, J. A. Roberts, N. Y. Nelson, C. J. Medforth, S. Jia, M. C. Simpson, K. M. Smith, J. Fajer, J. A. Shelnutt and D. Holten, J. Am. Chem. Soc., 1998, 120, 3781–3791 CrossRef CAS.
- X. Y. Zhang, E. C. Wasinger, A. Z. Muresan, K. Attenkofer, G. Jennings, J. S. Lindsey and L. X. Chen, J. Phys. Chem. A, 2007, 111, 11736–11742 CrossRef CAS.
- E. W. Findsen, J. A. Shelnutt, J. M. Friedman and M. R. Ondrias, Chem. Phys. Lett., 1986, 126, 465–471 CrossRef CAS.
- S. H. Courtney, T. M. Jedju, J. M. Friedman, L. Rothberg, R. G. Alden, M. S. Park and M. R. Ondrias, J. Opt. Soc. Am. B, 1990, 7, 1610–1614 CrossRef CAS.
- J. A. Shelnutt, C. J. Medforth, M. D. Berber, K. M. Barkigia and K. M. Smith, J. Am. Chem. Soc., 1991, 113, 4077–4087 CrossRef CAS.
- J. A. Shelnutt, S. A. Majumder, L. D. Sparks, J. D. Hobbs, C. J. Medforth, M. O. Senge, K. M. Smith, M. Miura, L. Luo and J. M. E. Quirke, J. Raman Spectrosc., 1992, 23, 523–529 CAS.
- W. Jentzen, E. Unger, G. Karvounis, J. A. Shelnutt, W. Dreybrodt and R. Schweitzer-Stenner, J. Phys. Chem., 1996, 100, 14184–14191 CrossRef CAS.
- X.-Z. Song, W. Jentzen, S.-L. Jia, L. Jaquinod, D. J. Nurco, C. J. Medforth, K. M. Smith and J. A. Shelnutt, J. Am. Chem. Soc., 1996, 118, 12975–12988 CrossRef CAS.
- S.-L. Jia, W. Jentzen, M. Shang, X.-Z. Song, J.-G. Ma, W. R. Scheidt and J. A. Shelnutt, Inorg. Chem., 1998, 37, 4402–4412 CrossRef CAS.
- D. H. Jeong, D. Kim, D. W. Cho and S. C. Jeoung, J. Raman Spectrosc., 2001, 32, 487–493 CrossRef CAS.
- Q. Huang, C. J. Medforth and R. Schweitzer-Stenner, J. Phys. Chem. A, 2005, 109, 10493–10502 CrossRef CAS.
- L. X. Chen, J. Electron Spectrosc. Relat. Phenom., 2001, 119, 161–174 CrossRef CAS.
- L. X. Chen, W. J. H. Jager, G. Jennings, D. J. Gosztola, A. Munkholm and J. P. Hessler, Science, 2001, 292, 262–264 CrossRef CAS.
- L. X. Chen, X. Zhang, E. C. Wasinger, K. Attenkofer, G. Jennings, A. Muresan and S. Lindsey Jonathan, J. Am. Chem. Soc., 2007, 129, 9616–9618 CrossRef CAS.
- K. M. Barkigia, M. W. Renner, L. R. Furenlid, C. J. Medforth, K. M. Smith and J. Fajer, J. Am. Chem. Soc., 1993, 115, 3627–3635 CrossRef CAS.
- L. Campbell, S. Tanaka and S. Mukamel, Chem. Phys., 2004, 299, 225–231 CrossRef CAS.
- T. S. Rush III, P. M. Kozlowski, C. A. Piffat, R. Kumble, M. Z. Zgierski and T. G. Spiro, J. Phys. Chem. B, 2000, 104, 5020–5034 CrossRef CAS.
- E. J. Baerends, G. Ricciardi, A. Rosa and S. J. A. van Gisbergen, Coord. Chem. Rev., 2002, 230, 5–27 CrossRef CAS.
- L. Campbell and S. Mukamel, J. Chem. Phys., 2004, 121, 12323–12333 CrossRef CAS.
- R. K. Pandey and S. Mukamel, Journal of Chemical Physics, 2006, 124, 094106-1–094106-10.
- C. J. Medforth, M. O. Senge, K. M. Smith, L. D. Sparks and J. A. Shelnutt, J. Am. Chem. Soc., 1992, 114, 9859–9869 CrossRef CAS.
- J. A. Shelnutt, J. Am. Chem. Soc., 1987, 109, 4169–4173 CrossRef CAS.
- J. L. Retsek, C. M. Drain, C. Kirmaier, D. J. Nurco, C. J. Medforth, K. M. Smith, I. V. Sazanovich, V. S. Chirvony, J. Fajer and D. Holten, J. Am. Chem. Soc., 2003, 125, 9787–9800 CrossRef CAS.
- J. Rodriguez and D. Holten, J. Chem. Phys., 1990, 92, 5944–5950 CrossRef CAS.
- A. V. Zamyatin, A. V. Gusev and M. A. J. Rodgers, J. Am. Chem. Soc., 2004, 126, 15934–15935 CrossRef CAS.
- L. X. Chen, Angew. Chem., Int. Ed., 2004, 43, 2886–2905 CrossRef CAS.
- L. X. Chen, Annu. Rev. Phys. Chem., 2005, 56, 221–254 CrossRef CAS.
- G. Jennings, W. J. H. Jaeger and L. X. Chen, Rev. Sci. Instrum., 2002, 72, 362–368 CrossRef.
- C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489–492 CrossRef CAS.
- M. Chergui and A. H. Zewail, ChemPhysChem, 2009, 10, 28–43 CrossRef CAS.
- M. Saes, C. Bressler, R. Abela, D. Grolimund, S. L. Johnson, P. A. Heimann and M. Chergui, Phys. Rev. Lett., 2003, 90, 047403–047403 CrossRef.
- G. te Velde, F. M. Bickelhaupt, S. J. A. van Gisbergen, C. Fonseca Guerra, E. J. Baerends, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, J.K.A., M. R. Pederson, D. J. Singh and F.C., Phys. Rev. B: Condens. Matter, 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- W. Jentzen, X. Z. Song and J. A. Shelnutt, J. Phys. Chem. B, 1997, 101, 1684–1699 CrossRef CAS.
- Y. Song, R. E. Haddad, S.-L. Jia, S. Hok, M. M. Olmstead, D. J. Nurco, N. E. Schore, J. Zhang, J. G. Ma, K. M. Smith, S. Gazeau, J. Pé caut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2005, 127, 1179–1192 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- L.-S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of the equilibrium constant for the ligation in the ground state of NiTMP. See DOI: 10.1039/c0sc00323a |
| This journal is © The Royal Society of Chemistry 2010 |