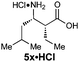
Unified synthesis of enantiopure β2h, β3h and β2,3-amino acids†‡
Shouyun
Yu
a,
Hiroshi
Ishida
a,
M. Elisa
Juarez-Garcia
b and
Jeffrey W.
Bode
*b
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA
bLaboratorium für Organische Chemie, ETH–Zürich, Zürich, 8093, Switzerland. E-mail: bode@org.chem.ethz.ch; Tel: +41 44 633 21 03
First published on 10th September 2010
Abstract
One for all, all for one! A single chiral auxiliary and synthetic route can be used for a three-step (cycloaddition, auxiliary removal and fragmentation) preparation of enantiopure β3h, β2h and β2,3-amino acids (>99% ee). This approach works for a wide variety of both natural and unnatural side chains, provides access to either enantiomeric form, and can be executed on a preparative scale without recourse to column chromatography.
Introduction
The discovery that oligomers of homologated amino acids form discrete, stable, and predictable secondary and tertiary structures has played a starring role in the emerging field of foldamers and the synthetic preparation of non-natural oligomers with defined structure and properties.1 Intense research into the preparation and properties of β-oligopeptides have led to remarkable achievements including the preparation of biologically active oligomers,2 artificial enzymes,3 and β-oligopeptides with tunable secondary, tertiary and quarternary structures.4 The enhanced structural and metabolic stability of these oligomers, relative to those composed from natural α-amino acids, make them one of the most promising approaches for the preparation of molecules with programmed properties and biological activities.Oligomers of various β-amino acids stand out as the most studied and sought after class of unnatural foldamers. The three general classes of the constituent β-amino acid monomers, as defined by Seebach,1b are the β3h-amino acids, the β2h-amino acids, and various diastereomers of β2,3-amino acids (Scheme 1). Unlike peptides consisting of natural α-amino acids, which are widely available in enantiomercially pure form at a nominal cost, each enantiopure β-amino acid monomer must be synthesized from a suitable chiral starting material or by asymmetric synthesis.5
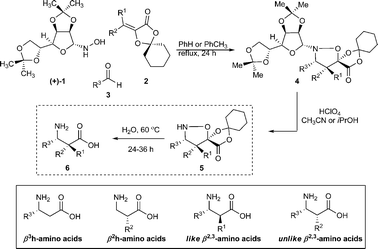 | ||
| Scheme 1 Three-step synthesis of enantiopure βh3, βh2, and β2,3-amino acids by isoxazolidine fragmentation. | ||
For enantiopure β3h-amino acids, Seebach and co-workers have refined the Arndt–Eistert homologation of α-amino acids with diazomethane. This method is widely employed and is the basis for the commercially available β3h-amino acids,6 but it is not well suited to the preparation of monomers with unnatural side chains or configurations due to the requirement of the enantiopure, Fmoc-protected α-amino acids as the starting material. The plethora of catalytic, enantioselective approaches to β-amino acids are rarely employed in practice due to issues of protecting groups and sub-optimal levels of enantioinduction,5 although several chiral auxiliary based routes are useful.7 To be amenable for the preparation of oligopeptides the monomers must be obtained in >99% ee as their Fmoc-protected β-amino acids.
Other important and desirable classes include β2,3-substituted variants and, especially, β2-amino acids, which are even more challenging to prepare and specialized methods are often needed for each different side chain. The contemporary challenge of reliable routes to diverse enantiopure β2-amino acid derivates has been beautifully outlined by Seebach in a recent review.8 The leading methods utilize several different types of chiral auxiliaries.9 Asymmetric hydrogenation of aminoacrylic acid derivatives10 or enantioselective Mannich-type reactions11 have proven useful in certain cases but with limited versatility. Some β2,3-amino acids are accessible by alkylation of β3h-derivatives12 or by enantioselective Michael addition of nitrogen nucleophiles to acrylate esters.13,14
In this communication we describe a general, unified approach to the preparation of β3h, β2h and both like- and unlike-β2,3-amino acids in enantiomerically pure form (>99% ee). This approach works for a wide variety of side chains from the corresponding aldehydes, provides access to either enantiomeric form, and can be executed on a preparative scale without recourse to column chromatography (Scheme 1). The key starting materials are readily available on scale at a reasonable cost, rendering this protocol an attractive alternative to β3h-amino acids and a viable approach to β2h and some β2,3-amino acids.
Results and discussion
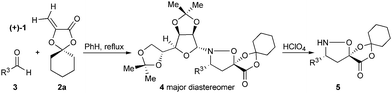
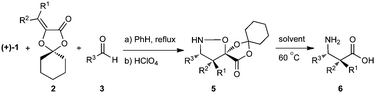
In the course of our studies on the synthesis of β3h-oligopeptides via chemoselective ligation of α-ketoacids and cyclic hydroxylamines,15 we recognized the potential for a variant of Vasella's asymmetric nitrone cycloaddition16 to provide general access to β-amino acid monomers via an unexpectedly facile N–O cleaving fragmentation reaction (Scheme 2). We investigated a number of different acrylates for the key nitrone cycloaddition and fragmentation and selected cyclohexanone-derived acrylate 2a for further studies.17 Refinement of this protocol identified advantages of D-gulose, rather than the original D-mannose, derived auxiliaries in terms of diastereoselection in the nitrone cycloaddition and increased crystallinity of the cycloadducts.18 Most importantly, the D-gulose-derived auxiliary afforded products that correspond with the “natural” configuration usually desired for β-oligopeptides. The D-mannose-derived auxiliary afforded the “unnatural” configuration; L-mannose is prohibitively expensive.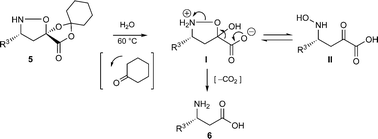 | ||
| Scheme 2 Fragmentation of isoxazolidine lactones to form β-amino acids. | ||
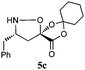
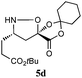
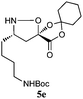
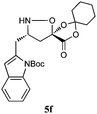
The cycloadducts were isolated with good to excellent levels of diastereoselectivity as solids that could be readily crystallized to high levels of diastereo- and enantiopurity (Table 1). Single crystal X-ray analysis confirmed the expected relative stereochemistry (see ESI‡), which follows the stereochemical model previously proposed by Vasella.19 The auxiliary could be removed with a variety of mild conditions, such as treatment with perchloric acid in acetonitrile, which allowed acid-sensitive protecting groups to survive. This two-step protocol was applied to several different aldehydes and the results are summarized in Table 1. Alternatively, the initial cycloadducts could be transformed prior to auxiliary removal, making possible the synthesis of precursors to a number of β3h-amino acids containing unnatural side chains (see Table 2 and ESI‡ for examples and procedures).
|
|
||||
|---|---|---|---|---|
| Entry | Isoxazolidine | dra | Yield (%)b | Yield (%)c |
| a dr of cycloadducts as determined by 1H NMR. b Isolated overall yield of cycloadducts; Number in parentheses refers to yield of enantiopure major diastereomer after recrystallization. c Isolated yield of isoxazolidines after auxiliary removal. d Overall yield for deprotection of N-Cbz and auxiliary removal. e Prepared from (−)−1. | ||||
| 1 |
|
87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:5:0 :8:5:0 |
93(56) | 73 |
| 2e |
|
75:18:7:0 |
93(47) | 83 |
| 3 |
|
86:9:5:0 |
96(59) | 58 |
| 4 |
|
84:8:8:0 |
69(43) | 68 |
| 5 |
|
83:11:6:0 |
95(50) | 47d |
| 6 |
|
86:8:6:0 |
88(55) | 47 |
| 7 |
|
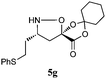
not determined | (71) | 71 |
| Entry | Isoxazolidines | Amino acids | Solvent | Yield (%)b |
|---|---|---|---|---|
| a All reactions performed with isoxazolidines or their HCl salts (0.1 M) in the indicated solvent (1 mL) at 60 °C 12–36 h. b Isolated yield of amino acids or their HCl salts. c Prepared from (+)−1. d Prepared from (−)−1. e Overall yield for fragmentation and Fmoc-protection. | ||||
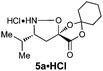
| 1c |
|
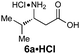
|
H2O | 100 |
| 2d |
|
|
H2O | 100 |
| 3c |
|
|
H2O/tBuOH | 92 |
| 4c |
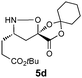
|
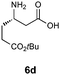
|
H2O/tBuOH | 93 |
| 5c |
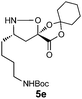
|
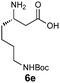
|
H2O/tBuOH | 96 |
| 6c |
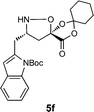
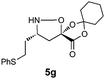
|
|
H2O/tBuOH | 42e |
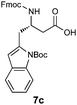
| 7c |
|
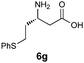
|
H2O/tBuOH | 70 |
| 8c |
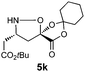
|
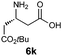
|
H2O/tBuOH | 84 |
| 9d |
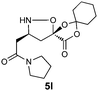
|
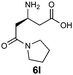
|
H2O/tBuOH | 97 |
| 10d |
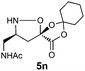
|
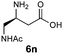
|
H2O/tBuOH | 99 |
| 11d |
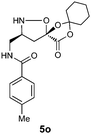
|
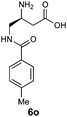
|
H2O/tBuOH | 87 |
Simply warming isoxazolidines 5 in water leads to spontaneous fragmentation to β-amino acids (Scheme 2). The only byproduct of this reaction is cyclohexanone, which does not interfere with the product isolation and excellent yields are usually obtained following precipitation or removal of solvent from the reaction mixture. Every isoxazolidine we have tested to date provides the corresponding enantiopure β3-amino acid in good to quantitative yield (Table 2). It was also possible to prepare Fmoc-protected amino acids by treatment of the crude reaction mixture with FmocCl (entry 6 and ESI‡).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amino acids | Acrylate | dra | Yield (%)b | Yield (%)c | Yield (%)d |
| a dr of cycloadducts as determined by 1H NMR. b Isolated yield of cycloadducts; number in parentheses refers to yield of enantiopure major diastereomer after recrystallization. c Isolated yield of isoxazolidines. d Isolated yield of amino acids. e The fragmentation of isoxazolidines performed in H2O/tBuOH. f The fragmentation of isoxazolidines performed in H2O. g The cycloaddition was carried out in toluene. | ||||||
| 1e |
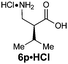
|
R1 = iPr | 77:23 |
85(50) | 92 | 100 |
| R2 = H | ||||||
| E-2d | ||||||
| 2e |
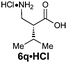
|
R1 = H | 84:16 |
91(62) | 94 | 100 |
| R2 = iPr | ||||||
| Z-2d | ||||||
| 3e |
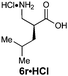
|
R1 = iBu | 85:15 |
88(61) | 91 | 99 |
| R2 = H | ||||||
| E-2f | ||||||
| 4e |
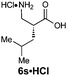
|
R1 = H | 84:16 |
90(57) | 92 | 100 |
| R2 = iBu | ||||||
| Z-2f | ||||||
| 5e |
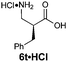
|
R1 = Bn | 75:25 |
88(47) | 90 | 99 |
| R2 = H | ||||||
| E-2e | ||||||
| 6e |
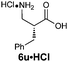
|
R1 = H | 82:18 |
91(63) | 79 | 98 |
| R2 = Bn | ||||||
| Z-2e | ||||||
| 7f |
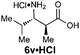
|
R1 = Me | 70:26:4:0 |
42(27) | 98 | 100 |
| R2 = H | ||||||
| E-2b | ||||||
| 8f |
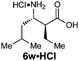
|
R1 = Et | 71:26:3:0 |
75(43) | 99 | 100 |
| R2 = H | ||||||
| E-2c | ||||||
| 9e,g |
|
R1 = H | not determined | 59(39) | 93 | 100 |
| R2 = Et | ||||||
| Z-2c | ||||||
Encouraged by these results for the synthesis of β3h-amino acids, we sought to extend this procedure to the preparation of more challenging β2h and β2,3-amino acids. The synthesis of the β2h-amino acid required the cycloaddition of a formaldehyde-derived nitrone with β-substituted acrylate 2, which can be prepared from the identical aldehydes used for the preparation of the β3-amino acids. Importantly, either the pure E or Z form of the acrylate can be accessed by known procedures from a phosphonate prepared in two steps from glyoxylic acid, cyclohexanone, and diethylphosphite.20 The cycloadditions were conducted by simply combining the aldehyde, acrylate, and chiral auxiliary in benzene or toluene and refluxing overnight (Table 3). Although the diastereoselectivities were slightly lower than those observed for the synthesis of the β3h-amino acids, we could isolate diastereomerically pure products in moderate to good yields. The E and Z acrylates provided diastereomeric cycloadducts that result in the opposite absolute configuration at the side chain position; thus, the D-gulose chiral auxiliary can be used to prepare both enantiomers of the β2h-amino acids (entries 1–6). When higher aldehydes were employed for the cycloaddition, either like or unlike 2,3-disubstituted isoxazolidines were obtained in good yield and diastereoselectivity (entries 7–9). In either series, removal of the chiral auxiliary was accomplished without difficulty by treatment with perchloric acid. The hydrolysis and fragmentation of these isoxazolidines to the β2h-amino acids proceeded cleanly and in high yield, although care must be taken to avoid epimerization (entries 1–6). Determination of the enantiopurity of the resulting β2h-amino acids derived from the free base isoxazolidines, analyzed as their Fmoc-protected derivatives, revealed that the products were isolated in only 80% ee for β2h-phenylalanine 6u and 90% ee for β2h-valine 6q. We eventually traced the epimerization to the fragmentation step, implicating the discrete intermediacy of II in the reaction (Scheme 2). After careful investigation, we found that epimerization could be almost completely suppressed by performing the reaction under acidic conditions. Addition of 2 equiv. HCl (aq.) or using HCl salts of isoxazolidines as precursors to β2-amino acids is sufficient to prevent epimerization. In our preliminary studies on the synthesis of β2,3-amino acids with this chemistry (entries 7–9), we were pleased that fragmentation of both the like and unlike-isoxazolidines proceeded smoothly to give the expected β2,3-amino acids in excellent yield.
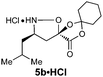
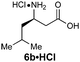
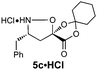
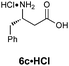
For the purpose of developing and optimizing this chemistry, we have isolated and characterized the two intermediates between the aldehyde starting material and the β-amino acids. The crystallinity of the gulose-derived isoxazolidines makes it possible to prepare enantipure β-amino acids without chromatography or isolation of the intermediates. For example, 1.7 g enantiopure (S)-β3h-phenylalanine 6c (>99% ee) was prepared from acrylate 2a and phenylacetaldehyde. Likewise, 2.2 g (R)-β2h-phenylalanine HCl salt 6u·HCl (99% ee) was prepared from acrylate Z-2e and paraformaldehyde (Scheme 3).
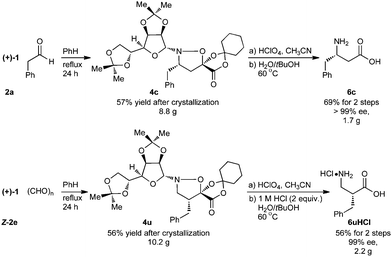 | ||
| Scheme 3 Preparative scale synthesis of enantiopure (S)-β3h-phenylalanine and (R)-β2h-phenylalanine without chromatography. | ||
Conclusions
In summary, we have documented a general, unified synthesis of enantiopure βh2 and βh3-amino acids from a single, readily available chiral auxiliary (Fig. 1). The same aldehyde starting material is the precursor to the amino acid side chain in either case, making possible the preparation of the valuable β-amino acid monomers by synthesis of the appropriate acrylate. The power of this route lies in its generality and reliability to afford enantiopure products. While other routes may be more direct for specific derivatives, this process may be widely applied to natural and unnatural derivatives of either configuration and without the need for dangerous or toxic reagents. We have also demonstrated that for both β2 and β3-amino acids it can be executed on a preparative scale without the need for column chromatography.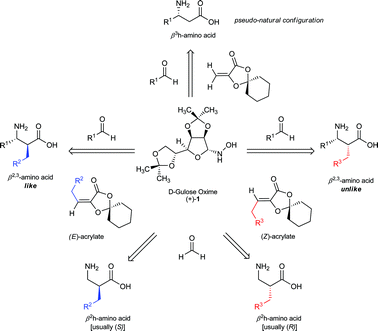 | ||
| Fig. 1 Overview of the synthesis of β-amino acids and their configuration from gulose-derived auxiliaries. The opposite enantiomers of those shown may be prepared from the L-gulose-derived auxiliary. | ||
Experimental section
Synthesis of (S)-β3h-phenylalanine (6c)
A mixture of phenyl acetaldehyde (4.0 mL, 34.3 mmol, 1.2 equiv.), acrylate 2a (5.73 g, 34.1 mmol, 1.2 equiv.) and D-gulose-derived hydroxylamine (+)−1 (7.82 g, 28.4 mmol, 1.0 equiv.) in PhH (140 mL) was heated to reflux with a Dean–Stark trap for 24 h. The solvent was removed under reduced pressure and the residue was crystallized from EtOAc (10 mL) and hexanes (30 mL) to give the diastereomerically pure cycloadduct (8.84 g, 57%) as white crystals.To a solution of this cycloadduct (7.78 g, 14.3 mmol, 1.0 equiv.) in CH3CN (140 mL) was added HClO4 (60%, 3.6 mL, 35.7 mmol, 2.5 equiv.) at RT and stirred 15 h. The reaction was quenched by the addition of aq. NaHCO3, extracted with EtOAc (3×), and the combined organic extracts were washed with brine and dried over Na2SO4. After concentration under reduced pressure, the residue was dissolved in 2:1 (v/v) H2O/tBuOH (150 mL) and stirred 24 h at 60 °C. The resulting mixture was cooled to RT, the solvent was removed and the remaining solid was collected and washed with EtOAc to give (S)-β3h-Phe (6c) (1.72 g, 69% for 2 steps, > 99% ee) as a light pale solid.
Synthesis of (R)-β2h-phenylalanine (6u•HCl)
A mixture of paraformaldehyde (1.29 g, 1.2 equiv.), acrylate Z-2e (10.29 g, 39.8 mmol, 1.2 equiv.) and D-gulose-derived hydroxylamine (+)−1 (9.20 g, 33.4 mmol, 1.0 equiv.) in PhH (150 mL) was heated to reflux with a Dean–Stark trap for 24 h. The solvent was removed under reduced pressure and the residue was crystallized from iPrOH (8 mL) and hexanes (24 mL) to give the diastereomerically pure cycloadduct (10.14 g, 56%) as white crystals.To a solution of this cycloadduct (10.14 g, 18.6 mmol, 1.0 equiv.) in CH3CN (140 mL) was added HClO4 (60%, 4.7 mL, 46.6 mmol, 2.5 equiv.) at RT and stirred 15 h. The reaction was quenched by the addition of aq. NaHCO3, extracted with EtOAc (3×), and the combined organic extracts were washed with brine and dried over Na2SO4. After concentration under reduced pressure, the residue was dissolved in 2:1 (v/v) H2O/tBuOH (150 mL) and concentrated aq. HCl (3.1 mL, 37.2 mmol, 2.0 equiv.) was added, then stirred 24 h at 60 °C. The resulting mixture was cooled to RT, the solvent was removed and the remaining solid was collected and washed with EtOAc to give (R)-β2h-Phe hydrochloride (6u·HCl) (2.23 g, 56% for 2 steps, 99% ee) as a light pale solid.
Acknowledgements
This work was supported by the Arnold and Mabel Beckman Foundation, the David and Lucille Packard Foundation, and Research Corporation (Cottrell Scholar Award to J. W. B.). H. I. was generously supported by Kyowa Kirin. We appreciate protocols from Nancy Carrillo, Justin Russak and Ying-Ling Chiang. We are grateful to BioBlocks, Inc. for a generous gift (+)-1.Notes and references
- For comprehensive reviews, see: (a) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS; (b) D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS; (c) M. I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, Org. Biomol. Chem., 2007, 5, 2884–2890 RSC; (d) W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS; (e) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS.
- (a) D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071 CrossRef CAS; (b) E. A. Porter, X. Wang, H.-S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS; (c) J. D. Sadowsky, M. A. Schmitt, H.-S. Lee, N. Umezawa, S. Wang, Y. Tomita and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 11966 CrossRef CAS; (d) Y. Imamura, N. Watanabe, N. Umezawa, T. Iwatsubo, N. Kato, T. Tomita and T. Higuchi, J. Am. Chem. Soc., 2009, 131, 7353–7359 CrossRef CAS.
- (a) X. J. Qiu, E. J. Petersson, E. E. Matthews and A. Schepartz, J. Am. Chem. Soc., 2006, 128, 11338–11339 CrossRef CAS; (b) D. S. Daniels, E. J. Petersson, X. J. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532–1533 CrossRef CAS; (c) E. J. Petersson, C. J. Craig, D. S. Daniels, X. J. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 5344–5345 CrossRef CAS; (d) M. M. Mueller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2009, 48, 922–925 CrossRef CAS.
- (a) R. P. Cheng and W. F. DeGrado, J. Am. Chem. Soc., 2001, 123, 5162–5163 CrossRef CAS; (b) P. I. Arvidsson, M. Rueping and D. Seebach, Chem. Commun., 2001, 649–650 RSC.
- For reviews, see: (a) Enantioselective Synthesis of β-Amino Acids (Ed: E. Juaristi), John Wiley & Sons: New York, 1997 Search PubMed; (b) Enantioselectiv e Synthesis of β-Amino Acids, 2nd ed. (Eds: E. Juaristi, V. A. Soloshonok), Wiley-VCH: New York, 2005 Search PubMed; (c) N. Sewald, Angew. Chem., Int. Ed., 2003, 42, 5794–5795 CrossRef CAS; (d) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991–8035 CrossRef CAS; (e) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656–1691 RSC.
- (a) D. Seebach, M. Overhand, F. N. M. Kuhnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef CAS; (b) G. Guichard, S. Abele and D. Seebach, Helv. Chim. Acta, 1998, 81, 187–206 CrossRef CAS.
- T. P. Tang and J. A. Ellman, J. Org. Chem., 2002, 67, 7819–7832 CrossRef CAS.
- D. Seebach, A. K. Beck, S. Capone, G. Deniau, U. Groselj and E. Zass, Synthesis, 2009, 1–32 CrossRef CAS.
- D. Seebach, L. Schaeffer, F. Gessier, P. Bindschadler, C. Jager, D. Josien, S. Kopp, G. Lelais, Y. Mahajan, P. Micuch, R. Sebesta and B. W. Schweizer, Helv. Chim. Acta, 2003, 86, 1852–1861 CrossRef CAS.
- For excellent reviews, see: (a) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278–1290 CrossRef CAS; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS For selected seminar reports, see:; (c) G. Zhu, Z. Chen and X. Zhang, J. Org. Chem., 1999, 64, 6907–6910 CrossRef CAS; (d) D. Heller, J. Holz, H.-J. Drexler, J. Lang and K. Drauz, J. Org. Chem., 2001, 66, 6816–6817 CrossRef CAS; (e) M. Yasutake, I. D. Gridnev, N. Higashi and T. Imamoto, Org. Lett., 2001, 3, 1701–1704 CrossRef CAS; (f) Y.-G. Zhou, W. Tang, W.-B. Wang, W. Li and X. Zhang, J. Am. Chem. Soc., 2002, 124, 4952–4953 CrossRef CAS; (g) Y. Hsiao, N. R. Rivera, T. Rosner, S. W. Krska, E. Njolito, F. Wang, Y. Sun, J. D. Armstrong III, E. J. J. Grabowski, R. D. Tillyer, F. Spindler and C. Malan, J. Am. Chem. Soc., 2004, 126, 9918–9919 CrossRef CAS.
- For an excellent review, see: (a) A. Cordova, Acc. Chem. Res., 2004, 37, 102–112 CrossRef CAS For selected seminar reports, see:; (b) B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS; (c) A. G. Wenzel and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 12964–12965 CrossRef CAS; (d) J. Song, Y. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 6048–6049 CrossRef CAS; (e) Y. Chi, E. P. English, W. C. Pomerantz, W. S. Horne, L. A. Joyce, L. R. Alexander, W. S. Fleming, E. A. Hopkins and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 6050–6055 CrossRef CAS.
- D. Seebach, S. Abele, K. Gademann, G. Guichard, T. Hintermann, B. Jaun, J. L. Mathews, J. V. Schreiber, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1998, 81, 932–982 CrossRef CAS.
- (a) S. G. Davies, N. M. Garrido, D. Kruchinin, O. Ichihara, L. J. Kotchie, P. D. Price, A. J. PriceMortimer, A. J. Russell and A. D. Smith, Tetrahedron: Asymmetry, 2006, 17, 1793–1811 CrossRef CAS; (b) S. G. Davies, A. D. Smith and P. D. Price, Tetrahedron: Asymmetry, 2005, 16, 2833–2891 CrossRef CAS.
- For other useful methods for the preparation of β-amino acids, see: (a) A. R. Minter, A. A. Fuller and A. K. Mapp, J. Am. Chem. Soc., 2003, 125, 6846–6847 CrossRef CAS; (b) A. A. Fuller, B. Chen, A. R. Minter and A. K. Mapp, Synlett, 2004, 1409–1413 CAS; (c) A. A. Fuller, B. Chen, A. R. Minter and A. K. Mapp, J. Am. Chem. Soc., 2005, 127, 5376–5383 CrossRef CAS; (d) H.-S. Lee, J.-S. Park, B. M. Kim and S. H. Gellman, J. Org. Chem., 2003, 68, 1575–1578 CrossRef CAS.
- N. Carrillo, E. A. Davalos, J. A. Russak and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 1452–1453 CrossRef CAS.
- (a) A. Vasella, Helv. Chim. Acta, 1977, 60, 1273–1295 CrossRef CAS . For other applications of carbohydrate-derived chiral auxiliaries for the synthesis of hydroxylamines and isoxazolidines, see; (b) R. Fassler, D. E. Frantz, J. Oetiker and E. M. Carreira, Angew. Chem., Int. Ed., 2002, 41, 3054–3056 CrossRef CAS; (c) K. Kasahara, H. Iida and C. Kibayashi, J. Org. Chem., 1989, 54, 2225–2233 CrossRef CAS.
- (a) For the synthesis of the acrylates, see: A. S. Shevchuk, V. A. Podgornova and B. F. Ustavshchikov, Rus. J. Org. Chem., 2001, 37, 741–743 Search PubMed; (b) See the ESI for an improved procedure.
- Both of the D- and L-gulose-derived auxiliaries are commercially available from BioBlocks, Inc. Alternatively, they can be prepared in three steps without chromatography from the corresponding gulonic-acid lactones, which are both available from a variety of suppliers for ∼1 euro/gram in lots of 100 grams.
- R. Huber and A. Vasella, Tetrahedron, 1990, 46, 33–58 CrossRef CAS.
- (a) U. Schmidt, J. Langner, B. Kirschbaum and C. Braun, Synthesis, 1994, 1138 CrossRef CAS; (b) See the ESI for an improved procedure.
Footnotes |
| † Dedicated to Professor Andrea Vasella. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization for all compounds and preparation of key reagents and auxiliaries. CCDC 763138–763140. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00317d |
| This journal is © The Royal Society of Chemistry 2010 |