Dissecting tunicamycin biosynthesis by genome mining: cloning and heterologous expression of a minimal gene cluster†‡
Filip J.
Wyszynski
a,
Andrew R.
Hesketh
b,
Mervyn J.
Bibb
*b and
Benjamin G.
Davis
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: ben.davis@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 275652
bDepartment of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. E-mail: mervyn.bibb@bbsrc.ac.uk; Fax: +44 (0)1603 450778; Tel: +44 (0)1603 450773
First published on 10th September 2010
Abstract
Tunicamycin nucleoside antibiotics were the first known to target the formation of peptidoglycan precursor lipid I in bacterial cell wall biosynthesis. They have also been used extensively as inhibitors of protein N-glycosylation in eukaryotes, blocking the biogenesis of early intermediate dolichyl-pyrophosphoryl-N-acetylglucosamine. Despite their unusual structures and useful activities, little is known about their biosynthesis. Here we report identification of the tunicamycin biosynthetic genes in Streptomyces chartreusis following genome sequencing and a chemically-guided strategy for in silico genome mining that allowed rapid identification and unification of an operon fractured across contigs. Heterologous expression established a likely minimal gene set necessary for antibiotic production, from which a detailed metabolic pathway for tunicamycin biosynthesis is proposed. These studies unlock a comprehensive and unusual toolbox of biosynthetic machinery with which to create variants of this important natural product, allowing possible improved understanding of the mode of action and facilitating future redesign. We anticipate that these results will enable the generation of altered specific inhibitors of diverse carbohydrate-processing enzymes, including improved targeting of lipid I biosynthesis.
Introduction
The tunicamycins are fatty acyl nucleoside antibiotics, first isolated from the soil actinomycete Streptomyces lysosuperificus in 1971 and later from Streptomyces chartreusis.1 Their structures consist of an unusual eleven carbon aminodialdose core (tunicamine) to which uracil and N-acetylglucosamine (GlcNAc) are anomerically attached, alongside a range of amide-linked unsaturated fatty acids (Figure 1).2 A number of related natural products belong to the tunicamycin family, namely streptovirudins,3 corynetoxins,4 MM19290,5 mycospocidin6 and antibiotic 24010.7 All share the conserved carbohydrate core and presumably have similar biosynthetic pathways, but the genes required for their production have not been identified. The tunicamycins are potent inhibitors of bacterial cell wall biosynthesis, targeting MraY, which catalyzes the formation of the key peptidoglycan precursor undecaprenyl-pyrophosphoryl-N-acetylmuramoyl pentapeptide (lipid I).8 Unfortunately, these antibiotics have not been used clinically due to cytotoxicity against mammalian cells, associated with inhibition of eukaryotic protein N-glycosylation. Specific binding to the active site of UDP-GlcNAc:dolichyl phosphate GlcNAc-1-phosphate transferase (GPT) blocks production of the lipid-linked precursor dolichyl-pyrophosphoryl-N-acetyl-glucosamine (Dol-PP-GlcNAc) and terminates asparagine-linked glycoprotein synthesis at the first committed step.9 This property has also led to the widespread use of tunicamycin as a crucial tool in the study of glycoproteins.10 A number of synthetic studies towards the tunicamycins have been published,11 with two full syntheses,12 and preliminary biosynthetic investigations with labeled precursors have suggested the metabolic origin of some parts of the molecule (Figure 1(b) and ESI† for further details).13 Despite this interest, the absence of a sequence for the tunicamycin gene cluster (or any part of it) has hindered understanding of the biosynthetic pathway and it has remained poorly defined, 40 years after tunicamycin's first isolation. Deciphering the biosynthesis of natural products active against peptidoglycan biosynthesis, such as vancomycin,14 teicoplanin,15 moenomycin16 and the β-lactams,17 has provided insights into their modes of action, allowing identification and specific tailoring of key structural motifs. In this report, these studies are now extended to the tunicamycins with the identification and heterologous expression of the biosynthetic gene cluster (tun cluster) from S. chartreusis. Bioinformatic analysis of these tun genes yields important information on the biogenesis of complex linkages present in the molecule, particularly the unique α,β-1,1-glycosidic linkage and an unusual tail-to-tail coupling of two monosaccharide building blocks through a C–C bond. Highly selective inhibitors of MraY as well as rationally designed inhibitors of other important carbohydrate-processing enzymes can now be envisaged, utilising the toolbox of biosynthetic machinery presented herein.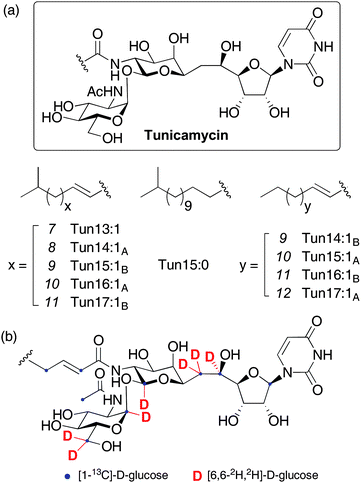 | ||
| Fig. 1 (a) Structures of the tunicamycins and (b) sites of label incorporation from prior feeding experiments with labelled D-glucose.13 See ESI† for a detailed interpretation of possible label incorporation pathways. | ||
Results
Identification of the tunicamycin biosynthetic gene cluster
Natural product gene clusters, particularly those for polyketide or non-ribosomal peptide biosynthesis, have often been identified by PCR amplification of highly conserved signature genes using degenerate primers, followed by screening of genomic libraries for the presence of these sequences.18 In contrast, tunicamycin is unlikely to require a large number of genes for its production and few of its biosynthetic genes can be predicted with enough precision to confidently assign a particular genetic homologue as a highly conserved probe sequence for degenerate primer design. For this reason, de novo genome scanning of a known tunicamycin producer together with ‘filtered’ genome mining was used instead as a rapid, more direct method to identify the genes for tunicamycin biosynthesis.High molecular weight genomic DNA was isolated from the two bacterial strains known to produce tunicamycin - S. chartreusis NRL3882 and S. lysosuperificus ATCC31396.13 The latter was shown to contain a high copy number plasmid and since this would bias subsequent sequencing data towards plasmid sequences, we elected to work with S. chartreusis NRL3882. The genome was sequenced to 36× coverage, generating 3112 contigs19 with a maximum size of 53.9 kb and an N50 average size20 of 4.6 kb. The contigs covered 7.95 Mb of the S. chartreusis chromosome with a G+C content of 70 mol%, consistent with existing data for Streptomyces genomes.21
Using bioinformatics tools tBLASTn22 and Artemis,23 these contigs were scanned for the presence of candidate tun genes. The tunicamycins contain many unique structural motifs (Figure 1) created by proteins with functions for which no similar examples or parallels are known. We therefore selected from a wide range of existing gene products with possible, chemically-similar function to putative members of the tunicamycin biosynthetic cluster. These focused on unique features and included: 1→1-linking glycosyltransferases (such as those from 3,3′-neotrehalosadiamine biosynthesis24 and from the OtsA-OtsB, TreY-TreZ and TreS pathways of trehalose biosynthesis25), N-acetyl-hexosamine N-deacetylases (e.g., from mycothiol, glycophosphatidylinositol, neomycin and teicoplanin biosynthesis26), lipid-processing proteins and numerous examples from the abundant NDP-hexose epimerase and dehydratase families. Using the combined presence of more than one of these reactivities (putative activity indicated by high similarity scores in tBLASTn to known enzyme activities) coupled with a knowledge of the required enzymology as a sifting strategy by which to mine the genome sequence data, a single contig was identified as the only ‘hit’ containing homologues of inosityl-GlcNAc deacetylase, hexose epimerase/dehydratase and acyl carrier protein encoded by a cluster of eleven open reading frames (ORFs). The 3′-end of this operon coincided with the end of the contig and terminated with the partial sequence of a putative GT-1 family glycosyltransferase (termed tunD), another key element of our reactivity filter. The full length sequences of glycosyltransferases homologous to TunD were screened against the S. chartreusis genome and an additional contig detected, containing the remaining portion of tunD and three further ORFs. In this way, the use of a bioinformatics filter, based on chemical logic, uniquely suggested the unification of an operon fractured across two distinct contigs. This unification was confirmed experimentally by generating a PCR product to bridge the gap between the two contigs, using primers matching the 5′ and 3′ ends of tunD and a genomic DNA template; its sequence confirmed that the two contigs were indeed adjacent on the chromosome and revealed that there were no bases between them. This joined contig contained 14 ORFs that appear to lie in a single operon, with many translationally coupled to the preceding gene (Figure 2). Analysis of the 5′ upstream region of each ORF in the cluster revealed that most had at least four bases of the GGAGG ribosomal binding site motif; regions preceding tunJ, tunM and tunN possess a GGA motif. Translational coupling to tunK likely allows effective expression of tunL and tunM. Further bioinformatic analysis revealed that these genes are likely to constitute a tunicamycin biosynthetic gene cluster and the predicted function of genes tunA-N and flanking ORFs are presented in Table 1 and Figure 2.
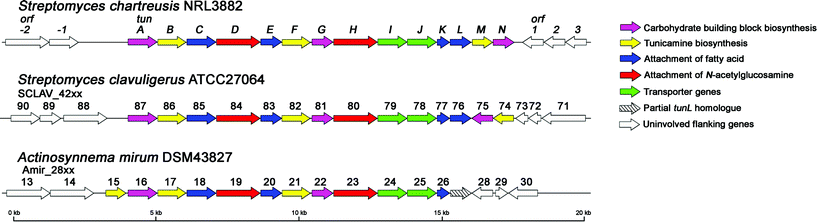 | ||
| Fig. 2 Genetic organisation of the tunicamycin biosynthetic gene cluster in S. chartreusis and its homologues in S. clavuligerus and A. mirum. | ||
| ORF | aaa | Proposed function in tunicamycin biosynthesis | Closest protein homologued, origin, (%Id/Si)b, Acc.c | Homologue in S. clavuligerus ATCC27064, aa, (%Id/Si), Acc. | Homologue in A. mirum DSM43827, aa, (%Id/Si), Acc. |
|---|---|---|---|---|---|
| a Number of amino acids. b (% Identity/Similarity). c Accession number. d Homologues from S. clavuligerus ATCC27064 and A. mirum DSM43827 were omitted from this column. | |||||
| ORF-2 | 406 | Integrase, Streptomyces sviceus ATCC 29083, (52/65), ZP_05020140 | |||
| ORF-1 | 297 | Transposase, Rhodococcus jostii RHA1, (50/64), YP_700005 | |||
| TunA | 321 | UDP-GlcNAc epimerase/dehydratase | NAD-dependent epimerase/dehydratase, Streptomyces sp. Mg1, (42/58), ZP_04996782 | SCLAV_4287, 276, (72/80), ZP_06773762 | Amir_2816, 322, (54/65), YP_003100592 |
| TunB | 338 | Uridine oxidoreductase | Radical SAM domain protein, Haloterrigena turkmenica DSM5511, (32/48), YP_003405396 | SCLAV_4286, 338, (90/95), ZP_06773761 | Amir_2817, 340, (78/87), YP_003100593 |
| TunC | 318 | N-Acyltransferase | GCN5-related N-Acetyltransferase, Fervidobacterium nodosum, (33/50), YP_001410548 | SCLAV_4285, 322, (60/72), ZP_06773760 | Amir_2818, 318, (43/57), YP_003100594 |
| TunD | 474 | Glycosyltransferase | Group 1 family glycosyltransferase, Thermococcus barophilus, (28/45), ZP_04876510 | SCLAV_4284, 461, (63/77), ZP_06773759 | Amir_2819, 451, (47/58), YP_003100595 |
| TunE | 234 | N-Deacetylase | GlcNAc-phosphatidylinositol de-N-acetylase, Cylindrospermopsis raciborskii, (43/57), ZP_06309433 | SCLAV_4283, 236, (77/85), ZP_06773758 | Amir_2820, 230, (63/76), YP_003100596 |
| TunF | 327 | UDP-GlcNAc- 4-epimerase | UDP-glucose 4-epimerase, Paenibacillus sp. oral taxon 786, (46/63), ZP_04852226 | SCLAV_4282, 327, (76/83), ZP_06773757 | Amir_2821, 332, (58/70), YP_003100597 |
| TunG | 203 | UMP phosphatase | Phosphoglycerate mutase, Frankia sp. CcI3, (29/47), YP_481446 | SCLAV_4281, 208, (65/73), ZP_06773756 | Amir_2822, 223, (50/60), YP_003100598 |
| TunH | 515 | UDP-tunicaminyluracil pyrophosphatase | Type I nucleotide pyrophosphatase, Burkholderia sp. 383, (35/50), YP_370731 | SCLAV_4280, 518, (66/76), ZP_06773755 | Amir_2823, 510, (53/65), YP_003100599 |
| TunI | 304 | ABC transporter ATP-binding subunit | Putative ABC transporter ATP-binding subunit, Streptomyces scabiei 87.22, (41/61), YP_003492364 | SCLAV_4279, 302, (77/88), ZP_06773754 | Amir_2824, 302, (60/73), YP_003100600 |
| TunJ | 262 | ABC transporter permease subunit | ABC-2 type transporter, Thermobaculum terrenum, (32/51), YP_003322218 | SCLAV_4278, 261, (76/83), ZP_06773753 | Amir_2825, 253, (61/78), YP_003100601 |
| TunK | 81 | Acyl carrier protein | Phosphopantetheine-binding protein, Catenulispora acidiphila DSM 44928, (32/61), YP_003117493 | SCLAV_4277, 81, (65/87), ZP_06773752 | Amir_2826, 79, (34/54), YP_003100602 |
| TunL | 229 | Phospholipid phosphatase | Phosphoesterase PA-phosphatase, Micromonospora aurantiaca, (33/42), ZP_06217896 | SCLAV_4276, 223, (52/67), ZP_06773751 | - |
| TunM | 216 | Radical SAM protein | Methyltransferase family protein, Saccharomonospora viridis, (48/63), YP_003133112 | SCLAV_4274, 212, (54/67), ZP_06773749 | Amir_2815, 232, (30/54), YP_003100591 |
| TunN | 152 | UTP pyrophosphatase | NUDIX hydrolase, Nakamurella multipartita, (36/55), YP_003200035 | SCLAV_4275, 170, (68/77), ZP_06773750 | - |
| ORF1 | 213 | Secreted protein, Streptomyces viridochromogenes, (81/90), ZP_05533938 | |||
| ORF2 | 573 | Secreted protein, Streptomyces viridochromogenes, (90/94), ZP_05533937 | |||
| ORF3 | 606 | Secreted protein, Streptomyces viridochromogenes, (82/89), ZP_05533936 |
Isolation and heterologous expression of the tun cluster
To confirm the involvement of the putative tun gene cluster in tunicamycin biosynthesis, it was introduced into Streptomyces coelicolor and the resulting recombinant strains screened for tunicamycin production. To do this, a cosmid library of S. chartreusis NRL3882 was assembled in Escherichia coli and probed with 32P-labelled PCR amplicons from within tunA and tunN, which are located at either terminus of the putative tun operon (Figure 2). Cosmids hybridizing to both PCR probes were isolated and restriction analysis revealed four that contained the entire 12 kb tun gene cluster positioned centrally in the cosmid insert. The backbones of these SuperCos1-based cosmids were subsequently exchanged for that of the conjugative and integrative vector pMJCOS127 through λ-RED-mediated recombination.28 These modified cosmids were transferred to S. coelicolor M1152 (see ESI†) by conjugation, and integration into the chromosomal ϕC31 phage attachment site achieved by selecting for apramycin resistance. In addition, a 12.9 kb SacI fragment from one of the four cosmids that contained the complete putative tun gene cluster plus 427 bp upstream of tunA and 500 bp downstream of tunN (neither of these additional DNA sequences is predicted to possess an entire ORF) was cloned into the conjugative and integrative vector pRT802.29 The resulting clone was similarly conjugatively transferred into an S. coelicolor M1146 host (for bacterial strain descriptions see ESI†). Heterologous expression of the putative tun gene cluster was monitored using an agar-diffusion bioassay; all five recombinant strains produced zones of inhibition when assayed against Bacillus subtilis, whereas control strains containing the relevant vectors alone did not (Figure 3A). This key observation shows that the tun cluster codes for a secondary metabolite with bactericidal activity against the B. subtilis reporter strain. To confirm the identity of this bactericidal metabolite, recombinant strains containing the putative tun gene cluster and the relevant controls were grown in liquid culture for five days and the pelleted mycelium extracted with methanol. LC/MS analysis of the putative tun-containing clones revealed a mass distribution and fragmentation pattern identical to that of tunicamycin; this metabolite was absent from the control strains (Figure 3B). The presence of tunicamycin was further confirmed by 1H NMR spectroscopy (see ESI†). The transfer of tunicamycin production to S. coelicolor M1146 by the SacI-cloned tun operon is particularly informative, since it likely delineates the boundaries of the tun gene cluster and defines the minimal biosynthetic gene cluster necessary for tunicamycin production and for future mechanistic and redesign studies.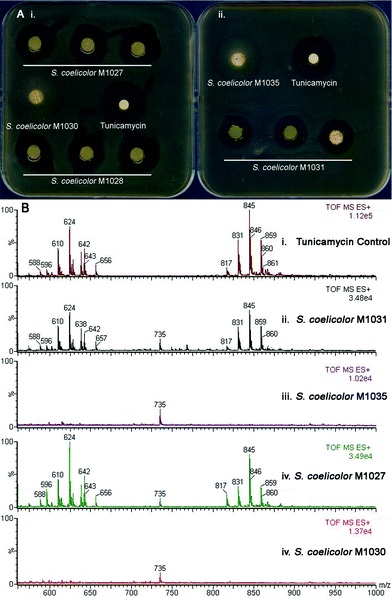 | ||
| Fig. 3 Evidence of heterologous production of tunicamycins in S. coelicolor. (A): Bioassay showing heterologous expression of (i) a genomic library-derived cosmid harboring the tun gene cluster introduced into a S. coelicolor M1152 host (giving recombinant strains S. coelicolor M1027 and M1028 derived from library cosmids 6N9 and 7C3, respectively) and control strain S. coelicolor M1030 (containing the same cosmid but without any insert sequence) and (ii) the minimal tun gene cluster cloned into pRT802 in S. coelicolor M1146 (giving recombinant strain S. coelicolor M1035) and control strain S. coelicolor M1031 (containing the empty pRT802 cosmid); (B): LC/MS analysis of (i) an authentic tunicamycin sample and mycelium extracts of these recombinant S. coelicolor strains (see ESI† for further chromatograms) (ii) M1031, (iii) M1035, (iv) M1027 and (v) M1030. See also Fig. S8, ESI† for 1H NMR analysis of the extracts. | ||
Discussion
Description of the tun gene cluster and identification of homologous clusters in Streptomyces clavuligerus and Actinosynnema mirum
The likely minimal tun gene cluster identified by heterologous expression comprises a contiguous 12.0 kb stretch of DNA containing a total of 14 ORFs, all of which are oriented in the same direction with many translationally coupled to the preceding gene, presumably to ensure appropriate levels of synthesis of each enzyme. This suggests that the entire cluster is contained within a single polycistronic transcript, which is feasible given its small overall size. The overall G+C content of this region is 65.0%, well below that of a typical Streptomyces genome or indeed the rest of the S. chartreusis genome. This suggests that the tun gene cluster was acquired from another, lower G+C content, organism at some point during its evolution.The ORFs flanking the proposed tun gene cluster are clearly not required for tunicamycin biosynthesis. The three flanking genes downstream of the tun cluster (ORF1-3) have close homologues in many Streptomyces genomes and encode conserved housekeeping genes. The two upstream flanking genes (ORF−1 and ORF−2) are homologous to transposase and integrase genes respectively, lending support to the hypothesis that S. chartreusis acquired the tun gene cluster by lateral gene transfer. The 1.9 kb region between ORF−1 and tunA contains a putative ORF with multiple frameshifts (“junk DNA”), again consistent with recent evolutionary acquisition.
Bioinformatic analysis revealed other potential tunicamycin producers. Homologous gene clusters were identified in A. mirum DSM4382730 and S. clavuligerus ATCC27064; the latter has been reported to produce the closely related antibiotic MM19290.5 Only minor differences were observed between the three gene clusters, suggesting a recently shared evolutionary heritage (Figure 2, Table 1).
Proteins encoded by the S. chartreusis gene cluster exhibit greatest similarity to those from S. clavuligerus, with amino acid sequence identities ranging from 52 to 90%. It is highly likely that genes annotated SCLAV_4274 to SCLAV_4287 are responsible for MM19290 biosynthesis in S. clavuligerus ATCC27064. Although the structure of MM19290 has not been reported, the high degree of homology with the tun genes from S. chartreusis strongly suggests that, like the streptovirudins and corynetoxins, this compound shares its core carbohydrate skeleton with tunicamycin.
Proteins encoded by the A. mirum gene cluster exhibit amino acid sequence identities with Tun proteins from S. chartreusis that range from 30 to 78%. While no full-length homologues of TunN or TunL were found, closer inspection of the A. mirum sequence revealed a truncated version of TunL that contained a number of frameshift mutations. Since this organism has not been reported to produce any antibiotics structurally related to the tunicamycins, it is probable that we have uncovered a silent gene cluster that has lost the ability to produce its tunicamycin-like metabolite.
Proposed biosynthetic pathway for the tunicamycins
Bioinformatic analysis of the tun gene cluster with BLAST and Artemis, together with identification of conserved active-site residues, allowed us to predict the functions of the products of tunA through to tunN (Table 1). Detailed descriptions of individual tun genes can be found in the ESI†. These assignments reconcile the genetic insight gained here with previous feeding experiments using labeled precursors,13 that together allow us to propose a detailed biosynthetic pathway to the tunicamycins (Figure 4). In our hypothesis, construction of the tunicaminyl-uracil core proceeds via the tail-to-tail coupling of uridine and galactosamine derivatives through a C–C linkage catalyzed by TunM. The involvement of UDP-4-keto-5,6-ene-GlcNAc (or another unsaturated substrate) as a substrate for TunM is supported by the presence of tunF and tunA, coding for a putative UDP-hexose-4-epimerase and a putative UDP-GlcNAc epimerase/dehydratase respectively and acting on UDP-GlcNAc, previously established as a metabolic precursor.13 The presence of two enzymes of similar/related function may suggest that since C-4 of the unsaturated substrate for TunM has lost all stereochemical information, its subsequent (or indeed prior) reduction after a coupling event may be enzymatically stereocontrolled. It may also be that in order to maintain no overall redox change, the product of TunA/F is an enol ether (as an alternative), raising the intriguing possibility of an enzymatic C–C bond forming reductive addition between enol ether and an aldehyde, a reaction that has no precedent in biology. Uridine-5′-aldehyde is also likely to feature as an intermediate and has been implicated in the biosynthesis of nikkomycin, polyoxin, liposidomycin and capuramycin nucleoside antibiotic families,31 although its formation and mechanistic role has not been studied in detail and remains poorly understood. For tunicamycin, we propose TunB-mediated formation, from uridine. TunB appears to be a radical SAM protein containing a 4Fe4S redox centre. We suggest that the requisite uridine is obtained from UTP by the sequential action of TunN (a nucleotide pyrophosphatase) and TunG (a nucleotide monophosphate phosphatase), respectively. It has also been suggested§ that UDP liberated halfway through the pathway by TunH followed by processing by TunN and TunG might also act as a source of uridine rather than cellular UTP; it should be noted that homologues of TunM are present and absent in the identified S. clavuligerus and A. mirum clusters, respectively. The coupling event may be mediated by TunB in combination with TunM, a methyltransferase homologue. Homologues of both of these enzymes catalyze radical processes, and thus the coupling of the two activated carbohydrate intermediates may proceed via a radical mechanism either by addition to an α,β-unsaturated ketone or through a Barbier-type mechanism. Alternatively, other (enol ether, uridine) substrates (see above) might be intermediates and suggest alternative reaction pathways, such as reductive enol ether-aldehyde coupling or uridyl radical addition to an enol ether or enone. It may also be that one enzyme alone (e.g. TunB) catalyzes this reaction. Regardless, this bond-forming process seems to be without precedent in nature. Subsequent tailoring of the pseudodisaccharide tunicaminyl-uracil core involves the formation of an α,β-1,1-trehalose linkage. Nucleotide-sugar pyrophosphatase TunH likely catalyzes the hydrolysis of UDP from this sugar for subsequent transfer of GlcNAc to the liberated anomeric position. This step we propose is catalyzed by a GT-1 family glycosyltransferase TunD, yielding the core pseudotrisaccharide skeleton of the tunicamycins with concurrent formation of two new stereocentres in the α,β-1,1-glycosidic bond. The final modification to this skeleton involves the introduction of a range of acyl chains to form each of the up to eighteen tunicamycin homologues that have been described.32 Since the heterologously expressed tun gene cluster produced fully acylated tunicamycins despite lacking a fatty acid synthase gene, the constituent acyl chains are most likely derived from the cellular pool of fatty acids, as previously observed in teicoplanin biosynthesis.33 The most likely function of TunL, a putative type 2 phosphatidic acid phosphatase (PAP2), is in the regulation of lipid synthesis in the producing bacterium. By down-regulating the levels of cellular phosphatidic acid and up-regulating levels of its cleavage product diacylglycerol, phospholipid biosynthesis is repressed and cellular pools of fatty acids can be diverted for use in tunicamycin biosynthesis via β-oxidative degradation pathways.34 Tunicamycin-producing organisms appear to have evolved an efficient way of perturbing the complex regulatory pathways of lipid metabolism regulation, allowing increased tunicamycin biosynthesis without negatively affecting vital cellular processes. Acyl carrier protein TunK next activates these sequestered fatty acids for subsequent acylation, presumably through the action of a fatty acyl-ACP ligase from primary metabolism, since no such ligase is present in the tun gene cluster. The tunicamycin core skeleton is prepared for amide bond formation by N-deacetylation with TunE, a putative member of the GlcNAc N-deacetylase family. TunC subsequently functions as an N-acyltransferase to install the sequestered and activated fatty acids, yielding the full range of tunicamycin homologues.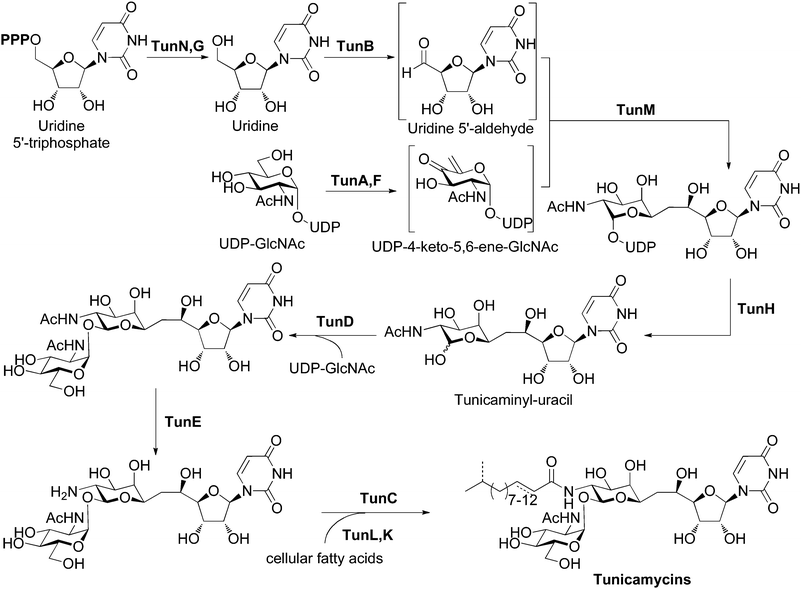 | ||
| Fig. 4 Proposed biosynthetic pathway for the tunicamycins. See also Fig. S2 and section S2, ESI† for a more detailed discussion of the roles of the tun gene products. | ||
The tun gene cluster described is relatively small in size, although a previous suggestion that as few as five genes would be necessary for the biosynthesis of tunicamycin has proved too conservative.35 Of the nine additional genes not originally predicted, two are involved in the generation of free uridine from UTP, contrary to suggestions that uridine would be obtained directly from primary metabolism.13 Two further genes are implicated in formation of UDP-tunicaminyl-uracil, one coding for a sugar epimerase supplementary to the dehydratase catalyzing UDP-4-keto-5,6-ene-GlcNAc formation, and one which potentially mediates the radical coupling event alongside the gene responsible for uridine oxidation. Hydrolysis of UDP from the undecose intermediate has also been suggested to require enzyme catalysis. Although the acyl side chains are likely to originate from cellular pools of fatty acids, consistent with the lack of a fatty acid synthase, the tun gene cluster still encodes two enzymes that may provide sufficient fatty acid flux and are involved in sequestering lipids and processing them prior to attachment. Finally, the last two additional tun genes are not directly involved in tunicamycin biosynthesis, but are likely to be crucial in conferring self-resistance to the producing organism. tunI and tunJ together encode for a putative ABC transporter, homologues of which are responsible for rapid ATP-driven efflux of antibiotics from cells in a large number of antibiotic-producing organisms.36
No regulatory genes were found in the tun gene cluster, suggesting that tunicamycin production may be subject to global control associated with growth rate reduction. The presence of rare TTA leucine codons (only 2% of S. coelicolor genes contain a TTA codon) in tunA and tunM may well reflect an element of translational regulation. In S. coelicolor, the accumulation of LeutRNAUUA is temporally regulated, and translation of mRNAs containing this codon may be largely confined to later stages of growth.37
Conclusions
With well over 8000 literature citations, the tunicamycins have attracted a great deal of attention for many years thanks to their unique structure and function and their potent and specific inhibition of N-acetyl-D-hexosamine-1-phosphate translocases involved in important cellular processes, particularly eukaryotic protein N-glycosylation and bacterial peptidoglycan biosynthesis. In this report we have identified the biosynthetic genes of a tunicamycin-family antibiotic for the first time, offering insights into the poorly understood biosynthetic pathway of this fascinating family of nucleoside antibiotics. Through molecular cloning and heterologous expression of the tun gene cluster in a S. coelicolor host, we have identified a minimal set of genes required for tunicamycin production. Additionally, we have identified close homologues of the tun gene cluster in A. mirum DSM43827 and S. clavuligerus ATCC27064. The latter organism is known to produce MM19290, an antibiotic closely related to tunicamycin, and based on the close similarity of its homologous cluster with the tun genes, we suggest that this cluster is likely responsible for MM19290 biosynthesis in S. clavuligerus ATCC27064. Furthermore, we propose that MM19290 shares the core structure of the tunicamycins and differs only in the nature of its acyl side chains. The availability of relatively inexpensive high throughput sequencing, combined with the genome scanning approach guided by the chemical logic described here provides a rapid and efficient way of identifying natural product gene clusters. The exponential increase in publicly available gene sequences in recent years has dramatically expanded the possibilities afforded by bioinformatic analysis. Our results suggest that such in silico mining of partially assembled genome sequences will constitute an increasingly effective tool during the early stages of dissecting a bacterial biosynthetic pathway.The findings presented here will allow detailed studies of tunicamycin biosynthesis. Functional characterization of individual enzymes will provide insight into how some of the unique linkages in tunicamycin are constructed. In addition, armed with this comprehensive toolbox of biosynthetic machinery, tunicamycin analogues with altered selectivity for bacterial MraY versus human GPT can now be sought, potentially leading to future therapeutic antibiotics with improved antibacterial activity and reduced cytotoxicity. Importantly, the mode of action of tunicamycin is orthogonal to all existing antibiotic drugs. Tunicamycin also provides a unique natural product template for inhibition of carbohydrate-processing enzymes. It represents a possible substrate and/or transition state mimic and hence transition state mimics of other important nucleotide sugar-dependent carbohydrate-processing enzymes might also be targeted by precursor-driven biosynthesis or chemoenzymatic methods, exchanging terminal functionalities of the tunicamycin structure.
Experimental
Materials and DNA manipulation methods
DNA manipulations were performed according to standard procedures for E. coli38 and Streptomyces.39 Unless otherwise stated, all Streptomyces media are listed in ref. 39 Chemical reagents, DNA oligonucleotides and media components were purchased from Sigma-Aldrich or BD Biosciences and used without further purification. Restriction endonucleases were purchased from New England Biolabs and remaining enzymes from Invitrogen.Bacterial strains, plasmids and culture conditions
The bacterial strains, plasmids and PCR primers used in this study are listed in Tables S1-3 (ESI†). All S. coelicolor strains were propagated on MS agar, S. chartreusis NRL3882 on OB agar and S. lysosuperificus ATCC31396 on MYM agar, at 30 °C (see ESI† for MYM and OB agar recipes). E. coli strains and B. subtilis EC1524 were routinely grown in Luria-Bertani broth (LB) or on 1.5% LB agar plates supplemented with appropriate antibiotics. For recombinant strain selection, antibiotics were used in the following concentrations: carbenicillin (100 µg/mL), kanamycin (50 µg/mL), apramycin (50 µg/mL), chloramphenicol (25 µg/mL) or nalidixic acid (25 µg/mL). For the isolation of genomic DNA, 10 µL of dense S. chartreusis or S. lysosuperificus spore preparations were inoculated into 50 mL TSB/YEME (1:1) in a 250 mL flask and incubated with shaking at 250 rpm and 30 °C for 24 h. For heterologous production, the growth medium was replaced with TYD13 and inoculated with spores from appropriate recombinant strains.Genome scanning of S. chartreusis NRL3882
Isolation of high molecular weight genomic DNA from S. chartreusis NRL3882 and S. lysosuperificus AATCC31396 was accomplished by published procedures.39 Genomic DNA S. chartreusis NRL3882 was sequenced and assembled by the University of Liverpool Advanced Genomics Facility using a Roche 454 Titanium pyrosequencing platform and the Roche Newbler (v2.0.00.20) assembler software. A BLAST database of the S. chartreusis genome was constructed and genome scanning, sequence analysis and functional annotation were performed using BLAST search tools22 and the Artemis v12.023 software package. A putative tun gene cluster was located spanning two non-overlapping contigs and the constituent genes were labeled tunA-N (Genbank accession code HQ172897). Using primers based on internal fragments from both ends of tunD, generation and sequencing of a PCR product spanning the gap between the two contigs showed their sequences to be contiguous on the bacterial chromosome.Generation and screening of S. chartreusis genomic library
Genomic DNA was partially digested with Sau3AI to yield 30–60 kb restriction fragments. Size-fractionated fragments were cloned into SuperCos1 (Stratagene), packaged using the Gigapack III Gold Packaging Extract Kit (Stratagene) and transduced into E. coli XL1 Blue MR (Stratagene), in each case following the manufacturers' instructions. At the Genome Analysis Centre (Norwich, UK), 3073 individual cosmid clones were picked and transferred to 96-well microtiter plates containing LB medium and ampicillin. DNA from these colonies was fixed onto nylon membrane filters according to published methods.38 Internal fragments of genes tunA and tunN were amplified by PCR from genomic DNA, labeled with 32P using the Rediprime II DNA Labelling System (Amersham) and used as hybridization probes for library screening. Eight clones hybridizing to both probes were subjected to restriction analysis with BamHI and XhoI, resulting in the selection of four cosmids (4H8, 5K7, 6N9 and 7C3) containing a complete, centrally located tun gene cluster.Preparation of recombinant S. coelicolor strains harbouring the tun gene cluster
The SuperCos1-derived library cosmids 4H8, 5K7, 6N9 and 7C3 were made conjugative and integrative by λ Red-mediated recombination of the vector sequence with a 5.2 kb SspI fragment from pMJCOS1 that contained an apramycin resistance cassette aac3(IV), oriT and ϕC31 integrase int and attachment site attB, as well as flanking sequences with identity to corresponding regions of the SuperCos1 backbone.40 The resultant constructs, named pIJ12315-8 (see Table S2, ESI†), were introduced into S. coelicolor M1152 via E. coli ET12567/pUZ8002 by conjugation according to published procedures41 and analysed for tunicamycin production. In addition, a 12.9 kb SacI fragment from cosmid 4H8 was cloned directly into the SacI site pRT802 yielding pIJ12003a. The SacI fragment contained the complete predicted tun gene cluster plus 427 bp upstream of tunA and 500 bp downstream of tunN; neither of these flanking DNA sequences is predicted to possess an entire ORF. Subsequently, pIJ12003a was conjugated into S. coelicolor M1146 via triparental mating using E. coli S17-1 and E. coli ET12567/pUZ8002.Analysis of tunicamycin production by recombinant S. coelicolor strains
Recombinant S. coelicolor M1025-1028, M1031 and their vector-only control strains were grown on R5 agar at 30 °C for 48 h. Agar cores were transferred to empty plates, which were then flooded with 50 °C soft nutrient agar inoculated with tunicamycin-sensitive B. subtilis EC1524. Additionally, sterile filter disks spotted with 15 µL of tunicamycin stock solution (1 mg/mL) were placed atop the solidified agar. The plates were grown at 30 °C for 18 h and examined for growth inhibition of the reporter strain. Furthermore, recombinant strains were grown in liquid TYD medium for 5 days at 30 °C and the suspected tunicamycin metabolites were isolated and characterized by methanolic mycelium extraction and LC/MS analysis, as described previously13 using a Micromass LCT (ESI-TOF MS) coupled to an Agilent 1200 Series LC System. Crude extracts were purified by flash chromatography using Fluka Kiegselgel 60 220–440 mesh silica gel (mobile phase: water/isopropanol/ethyl acetate 1:3:6) and subjected to 1H NMR spectroscopy along with an authentic sample to further confirm the presence of tunicamycin.Acknowledgements
We gratefully acknowledge Dr Govind Chandra for construction of the S. chartreusis NRL3882 BLAST database and Dr Juan-Pablo Gomez-Escribano for supplying S. coelicolor M1146 and M1152 (both at John Innes Centre, Norwich, UK). This work was supported by the EPSRC (DTA studentship to F. J. W.) and BBSRC (A. R. H. and M. J. B). B. G. D. is a Royal Society-Wolfson Research Merit Award recipient and is supported by an EPSRC LSI platform grant.References and notes
- (a) G. Tamura, Tunicamycin, Japan Scientific Societies Press, Tokyo, 1982 Search PubMed; (b) R. G. Hamill, U. S. Patent, 4237225, 1980 Search PubMed; (c) A. Takatsuki, K. Arima and G. Tamura, J. Antibiot., 1971, 24, 215 CAS.
- A. Takatsuki, K. Kawamura, M. Okina, Y. Kodama, T. Ito and G. Tamura, Agric. Biol. Chem., 1977, 41, 2307 CAS.
- H. Thrum, K. Eckardt, G. Bradler, R. Fugner, E. Tonew and M. Tonew, J. Antibiot., 1975, 28, 514 CAS.
- P. Vogel, D. S. Petterson, P. H. Berry, J. L. Frahn, N. Anderton, P. A. Cockrum, J. A. Edgar, M. V. Jago, G. W. Lanigan, A. L. Payne and C. C. J. Culvenor, Aust. J. Exp. Biol. Med. Sci., 1981, 59, 455 Search PubMed.
- M. Kenig and C. Reading, J. Antibiot., 1979, 32, 549 CAS.
- S. Nakamura, M. Arai, K. Karasawa and H. Yonehara, J. Antibiot., 1957, 10, 248 CAS.
- M. Mizuno, Y. Shimojima, T. Sugawara and I. Takeda, J. Antibiot., 1971, 24, 896 CAS.
- (a) G. Tamura, T. Sasaki, M. Matsuhashi, A. Takatsuki and M. Yamasaki, Agr. Biol. Chem., 1976, 40, 447 CAS; (b) P. E. Brandish, K. Kimura, M. Inukai, R. Southgate, J. T. Lonsdale and T. D. H. Bugg, Antimicrob. Agents. Chemother., 1996, 40, 1640 CAS.
- A. Heifetz, R. W. Keenan and A. D. Elbein, Biochemistry, 1979, 18, 2186 CrossRef CAS.
- A. D. Elbein, Trends Biochem. Sci., 1981, 6, 219 CrossRef CAS.
- (a) W. Karpiesiuk and A. Banaszek, Carbohydr. Res., 1997, 299, 245 CrossRef CAS; (b) S. J. Danishefsky, S. L. Deninno, S. Chen, L. Boisvert and M. Barbachyn, J. Am. Chem. Soc., 1989, 111, 5810 CrossRef CAS; (c) S. Ichikawa and A. Matsuda, Nucleosides, Nucleotides Nucleic Acids, 2004, 23, 239 CrossRef CAS; (d) F. Sarabia, L. Martin-Ortiz and F. J. Lopez-Herrera, Org. Biomol. Chem., 2003, 1, 3716 RSC; (e) J. Ramza and A. Zamojski, Tetrahedron, 1992, 48, 6123 CrossRef CAS.
- (a) A. G. Myers, D. Y. Gin and D. H. Rogers, J. Am. Chem. Soc., 1994, 116, 4697 CrossRef CAS; (b) T. Suami, H. Sasai, K. Matsuno and N. Suzuki, Carbohydr. Res., 1985, 143, 85 CrossRef CAS.
- B. C. Tsvetanova, D. J. Kiemle and N. P. J. Price, J. Biol. Chem., 2002, 277, 35289 CrossRef CAS.
- A. A. van Wageningen, P. N. Kirkpatrick, D. H. Williams, B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. M. Jones and P. J. Solenberg, Chem. Biol., 1998, 5, 155 CrossRef.
- T. L. Li, F. L. Huang, S. F. Haydock, T. Mironenko, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2004, 11, 107 CrossRef CAS.
- B. Ostash, E. H. Doud, C. Lin, I. Ostash, D. L. Perlstein, S. Fuse, M. Wolpert, D. Kahne and S. Walker, Biochemistry, 2009, 48, 8830 CrossRef.
- Y. Aharonowitz, G. Cohen and J. F. Martin, Annu. Rev. Microbiol., 1992, 46, 461 CrossRef CAS.
- T. Weber, K. Welzel, S. Pelzer, A. Vente and W. Wohlleben, J. Biotechnol., 2003, 106, 221 CrossRef CAS.
- R. Staden, Nucleic Acids Res., 1980, 8, 3673 CAS.
- The N50 average contig size represents the largest contig E such that at least half of the total size of the contigs is contained in contigs larger than E. For example in a collection of contigs with sizes 7, 4, 3, 2, 2, 1, and 1 kb (total size = 20 kb), the N50 length is 4 because we can cover 10 kb with contigs bigger than 4kb.
- Data for the size and overall G+C content of publicly available Streptomyces genomes (including both complete and draft assemblies) were obtained from the NCBI Genome Database (http://www.ncbi.nlm.nih.gov/sites/genome) and the Streptomyces Annotation Server (http://strepdb.streptomyces.org.uk/). Chromosome sizes of completed genomes ranged from 6.76 to 10.15 Mb, and G+C content ranged from 69 to 72%.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389 CrossRef CAS.
- K. Rutherford, J. Parkhill, J. Crook, T. Horsnell, P. Rice, M.-A. Rajandream and B. Barrell, Bioinformatics, 2000, 16, 944 CrossRef CAS.
- T. Inaoka and K. Ochi, J. Bacteriol., 2007, 189, 65 CrossRef CAS.
- K. A. L. De Smet, A. Weston, I. N. Brown, D. B. Young and B. D. Robertson, Microbiology, 2000, 146, 199 CAS.
- (a) N. Nakamura, N. Inoue, R. Watanabe, M. Takahashi, J. Takeda, V. L. Stevens and T. Kinoshita, J. Biol. Chem., 1997, 272, 15834 CrossRef CAS; (b) G. L. Newton, Y. Av-Gay and R. C. Fahey, J. Bacteriol., 2000, 182, 6958 CrossRef CAS; (c) A. W. Truman, L. Robinson and J. B. Spencer, ChemBioChem, 2006, 7, 1670 CrossRef CAS; (d) K. Yokoyama, Y. Yamamoto, F. Kudo and T. Eguchi, ChemBioChem, 2008, 9, 865 CrossRef CAS.
- S. Boakes, J. Cortes, A. N. Appleyard, B. A. M. Rudd and M. J. Dawson, Mol. Microbiol., 2009, 72, 1126 CrossRef CAS.
- (a) K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640 CrossRef CAS; (b) B. Gust, G. Chandra, D. Jakimowicz, Y. Q. Tian, C. J. Bruton and K. F. Chater, Adv. Appl. Microbiol., 2004, 54, 107 Search PubMed.
- M. A. Gregory, R. Till and M. C. M. Smith, J. Bacteriol., 2003, 185, 5320 CrossRef CAS.
- M. Land, A. Lapidus, S. Mayilraj, F. Chen, A. Copeland, T. G. D. Rio, M. Nolan, S. Lucas, H. Tice, J.-F. Cheng, O. Chertkov, D. Bruce, D. Bruce, L. Goodwin, S. Pitluck, M. Rohde, M. Göker, A. Pati, N. Ivanova, K. Mavromatis, A. Chen, K. Palaniappan, L. Hauser, Y.-J. Chang, C. D. Jeffries, T. Bretting, J. C. Detter, C. Han, P. Chain, B. J. Tindall, J. Bristow, J. A. Eisen, V. Markowitz, P. Hugenholtz, N. C. Kyripides and H.-P. Klenk, Standards in Genomic Sciences, 2009, 1, 46 Search PubMed.
- (a) L. Kaysser, L. Lutsch, S. Siedenberg, E. Wemakor, B. Kammerer and B. Gust, J. Biol. Chem., 2009, 284, 14987 CrossRef CAS; (b) L. Kaysser, S. Siedenberg and B. Gust, ChemBioChem, 2010, 11, 191 CrossRef CAS; (c) T. Ohnuki, Y. Muramatsu, S. Miyakoshi, T. Takatsu and M. Inukai, J. Antibiot., 2003, 56, 268 CAS; (d) M. Winn, R. J. M. Goss, K.-I. Kimura and T. D. H. Bugg, Nat. Prod. Rep., 2010, 27, 279 RSC.
- B. C. Tsvetanova and N. P. J. Price, Anal. Biochem., 2001, 289, 147 CrossRef CAS.
- A. Borghi, D. Edwards, L. F. Zerilli and G. C. Lancini, J. Gen. Microbiol., 1991, 137, 587 CAS.
- G. M. Carman and G. S. Han, J. Biol. Chem., 2009, 284, 2593 CAS.
- N. P. J. Price and B. Tsvetanova, J. Antibiot., 2007, 60, 485 CrossRef CAS.
- C. Mendez and J. A. Salas, Res. Microbiol., 2001, 152, 341 CrossRef CAS.
- (a) K. F. Chater, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 761 CrossRef CAS; (b) B. K. Leskiw, R. Mah, E. J. Lawlor and K. F. Chater, J. Bacteriol., 1993, 175, 1995 CAS.
- J. Sambrook and D. Russell, Molecular Cloning: A Laboratory Manual, 3rd edn., Cold Spring Harbor Laboratory Press, New York, 2000 Search PubMed.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Practical Streptomyces Genetics, John Innes Foundation, Norwich, 2000 Search PubMed.
- K. Yanai, T. Murakami and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9661 CrossRef CAS.
- B. Gust, T. Kieser and K. F. Chater, REDIRECT technology: PCR-targeting system in Streptomyces coelicolor, John Innes Foundation, Norwich, 2002 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of individual tun genes, a summary of prior studies into tunicamycin biosynthesis, experimental procedures, chromatograms and NMR spectra. See DOI: 10.1039/c0sc00325e/ |
| ‡ The sequences reported in this publication for the tun cluster are available in the Genbank database, accession code HQ172897. |
| § We thank a referee for this very useful suggestion. |
| This journal is © The Royal Society of Chemistry 2010 |