Highly enantioselective synthesis of 1,3-bis(hydroxymethyl)-2-oxindoles from unprotected oxindoles and formalin using a chiral NdIII complex†
Ke
Shen
,
Xiaohua
Liu
,
Wentao
Wang
,
Gang
Wang
,
Weidi
Cao
,
Wei
Li
,
Xiaolei
Hu
,
Lili
Lin
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, P. R. China. E-mail: xmfeng@scu.edu.cn; Fax: + 86 28 85418249; Tel: + 86 28 85418249
First published on 21st September 2010
Abstract
Highly enantioselective synthesis of 1,3-bis(hydroxymethyl)-2-oxindoles was established through chiral NdIII-N,N′-dioxide in up to 93% yield and >99% ee under mild conditions. The key features of this approach include using cheap and readily available formalin as a hydroxymethylation C1 unit and unprotected 3-substituted-2-oxindoles as nucleophiles directly. The initial C-aldol product from 3-substituted-2-oxindole was converted to the corresponding 1,3-bis(hydroxymethyl)-2-oxindole derivative immediately under the reaction conditions. Moreover, the catalyst was water-tolerant, and ligand and Nd(OTf)3 could be easily recovered and reused with no loss in catalytic activity and enantioselectivity. The synthetic utility of this methodology was accomplished with the synthesis of key intermediates of physostigmine and coerulescine.
Introduction
In recent years, great effort has been devoted to the hydroxymethylation reaction, which represent one of the most powerful and atom-economical one-carbon extension methods.1 In this regard, catalytic asymmetric hydroxymethylations of silyl enolates,2 ketones,3 α-cyanopropionates4 and β-keto esters5 have been intensively studied in the past decade. Most recently, Kobayashi and co-workers tried one example of asymmetric hydroxymethylation of N-Boc-3-methyl-2-oxindole with formalin (37% aqueous formaldehyde solution) using a chiral bipyridine-ScIII complex, delivering the desired product in 59% yield with 82% ee.3c Later, Yuan et al. reported a bifunctional thiourea-catalyzed asymmetric hydroxymethylation of N-Boc-oxindoles using paraformaldehyde, giving adducts in 80–99% yield with 5–91% ee.3d In view of the importance of oxindoles with potential medicinal interest and unique biological activities,6 the hydroxymethylation of oxindoles is therefore considered to be very important and urgent.However, the synthesis of N-Boc-oxindoles usually involves the addition of Grignard reagents to isatin derivatives, N-protecting and Pd/C reduction. The preparation features of this substrate as well as the deprotection of Boc group in the subsequent derivation reactions potentially limit the practical application. In contrast, the unprotected 3-substituted-2-oxindoles, which are commercially available or could be prepared directly from 2-oxindole under mild conditions, provide a good way to compensate for this limitation.7 On the other hand, there are still challenges associated with the use of formalin (the most convenient source of formaldehyde) as a useful C1 unit in Lewis acid catalysis due to its high reactivity and the water contained. To this point, the water-tolerant nature of rare earth metals (REs) make them potentially appropriate candidates for this reaction.8 Moreover, RE(OTf)3 might be facile for recovery from the aqueous layer during the workup step.8a Herein, to achieve protecting-group-free synthesis,6d we wish to address these issues and describe a general, practical, and highly enantioselective synthesis of 1,3-bis(hydroxymethyl)-2-oxindoles with formalin, in up to 93% yield and >99% ee, using a recyclable NdIII-N,N′-dioxide complex.9
It was rationalized that the addition between 1 and formaldehyde was likely to occur (Scheme 1(a)) through an in situ generated O-bond NdIII-enolate.10 To our great surprise, the reaction proceeded smoothly and the major product was 1,3-bis(hydroxymethyl)-2-oxindole 311 even when only 1.0 equiv. formalin was used. Interestingly, the isolated small amount of C-aldol product 212 could be further converted to N-aldol product 3 immediately in the presence of NdIII-N,N′-dioxide complex.13 This method could be a valuable expansion to the previously reported base-catalyzed N-hydroxymethylation reaction,14 due to the N-hydroxymethyl derivatives of nitrogen heterocycles as possible prodrugs.15
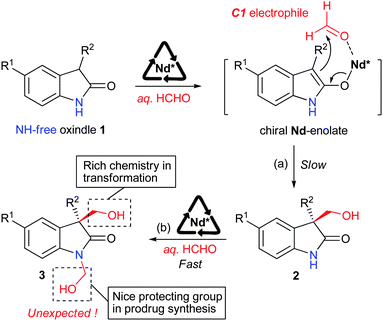 | ||
| Scheme 1 Strategy and proposed activation model; Nd* = chiral neodymium complex. | ||
Results and discussion
Initially, we probed the optimal reaction conditions using commercially available unprotected oxindole 1a and 1.0 equivalent formalin as model substrates. The reaction proceeded sluggishly and the major product was 1,3-bis(hydroxymethyl)-2-oxindole 3a, which had the same ee as that of 3-hydroxymethyl-2-oxindole 2a. It should be mentioned that the C-aldol adduct 2a could be transformed to 3a very rapidly in the presence of formalin. On the other hand, no single N-aldol product was observed. Thus, the initial C-aldol addition step was identified to be the rate-determining-step. Considering that the N-hydroxymethylated moiety of 3a is a good protecting group in prodrug synthesis, we then increased the amount of formalin to 2.0 equivalents to improve the yield of 3a. The combination of Sc(OTf)3 and N,N′-dioxide L3, derived from L-ramipril, delivered 3a in 46% yield and 55% ee (Table 1, entry 3).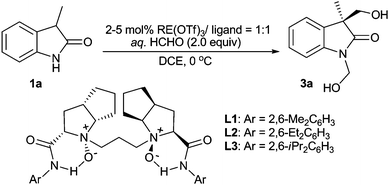
| Entrya | RE(OTf)3 | L | r RE3+/Åb | Lewis acidity of RE3+ | Yield (%)c | ee (%)d |
|---|---|---|---|---|---|---|
| a Unless noted otherwise, the reactions were conducted on 0.1 mmol scale, 5 mol% catalyst in 0.6 mL DCE for 36 h. b Ionic radii; see ref. 8. c Isolated yield. d Determined by chiral HPLC analysis. e On 10 mmol scale, 2 mol% L3-Nd(OTf)3 for 36 h. Both Nd(OTf)3 and L3 were recovered. f 5 mol% of recovered L3 and Nd(OTf)3 were used. g 3 Å MS was added (3 mg). | ||||||
| 1 | Sc(OTf)3 | L1 | 0.754 |
|
20 | 8 |
| 2 | Sc(OTf)3 | L2 | 0.754 | 28 | 26 | |
| 3 | Sc(OTf)3 | L3 | 0.754 | 46 | 55 | |
| 4 | Lu(OTf)3 | L3 | 0.861 | 50 | 33 | |
| 5 | Yb(OTf)3 | L3 | 0.868 | 72 | 40 | |
| 6 | Ho(OTf)3 | L3 | 0.901 | 65 | 70 | |
| 7 | Dy(OTf)3 | L3 | 0.912 | 70 | 68 | |
| 8 | Tb(OTf)3 | L3 | 0.923 | 60 | 70 | |
| 9 | Sm(OTf)3 | L3 | 0.958 | 88 | 83 | |
| 10 | Nd(OTf) 3 | L3 | 0.983 | 83 | 87 | |
| 11 | Pr(OTf)3 | L3 | 0.99 | 82 | 84 | |
| 12 | La(OTf)3 | L3 | 1.032 | 90 | 78 | |
| 13e | Nd(OTf)3 | L3 | 0.983 | 75 | 86 | |
| 14f | Nd(OTf)3 | L3 | 0.983 | 82 | 87 | |
| 15g | Nd(OTf)3 | L3 | 0.983 | 80 | 89 |
Ligands L1 and L2 with less sterically hindered substituents at the phenyl ring afforded much less satisfactory results (8% and 26% ee, respectively; Table 1, entries 1 and 2). When different RE(OTf)3 salts were evaluated, some phenomena were noteworthy. The enantiomeric excess of 3a was significantly influenced by slight differences in ionic radii and Lewis acidity of the REs.8b Notably, the reaction was most effectively promoted by the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Nd(OTf)3/L3 complex in CH2ClCH2Cl (DCE) at 0 °C, delivering product 3a in 83% yield with 87% ee (Table 1, entry 10), better than the other RE sources (Table 1, entries 3–9, 11-12). Typically, Sc(OTf)3, Yb(OTf)3 and Lu(OTf)3, which have smaller ionic radii and stronger Lewis acidities among the REs, generally gave lower enantioselectivities (Table 1, entries 3–5). Gratifyingly, catalyst loading could be reduced to 2 mol% on a gram-scale reaction, with marginal loss of yield and enantiomeric excess (Table 1, entry 13). It should be noted that Nd(OTf)3 was easily recovered from the aqueous phase by extraction, and optically pure ligand L3 was recovered through chromatography on silica gel (Table 1, entry 13). Ligand L3 and Nd(OTf)3 could be reused without any loss in catalytic activity and enantioselectivity (Table 1, entry 14). When 3 Å MS was added, the reaction rate was faster with a slight increase in ee (Table 1, entry 15).
:1 Nd(OTf)3/L3 complex in CH2ClCH2Cl (DCE) at 0 °C, delivering product 3a in 83% yield with 87% ee (Table 1, entry 10), better than the other RE sources (Table 1, entries 3–9, 11-12). Typically, Sc(OTf)3, Yb(OTf)3 and Lu(OTf)3, which have smaller ionic radii and stronger Lewis acidities among the REs, generally gave lower enantioselectivities (Table 1, entries 3–5). Gratifyingly, catalyst loading could be reduced to 2 mol% on a gram-scale reaction, with marginal loss of yield and enantiomeric excess (Table 1, entry 13). It should be noted that Nd(OTf)3 was easily recovered from the aqueous phase by extraction, and optically pure ligand L3 was recovered through chromatography on silica gel (Table 1, entry 13). Ligand L3 and Nd(OTf)3 could be reused without any loss in catalytic activity and enantioselectivity (Table 1, entry 14). When 3 Å MS was added, the reaction rate was faster with a slight increase in ee (Table 1, entry 15).
To evaluate the water-tolerant capability of L3-Nd(OTf)3 the hydroxymethylation reaction was conducted with 2.0 equiv. formalin and additional water. As shown in Fig. 1, the reaction could tolerate 30 equiv. of water although the amount of water influenced the yield and enantioselectivity to some extent. Introduction of surfactants such as CH3(CH2)11SO3Na or tetrabutylammonium chloride decreased the ee sharply.16
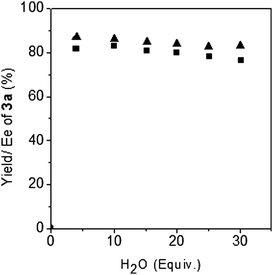 | ||
| Fig. 1 Water-tolerance experiment. Reactions performed with 0.1 mmol 1a, 2.0 equivalent formalin, 5 mol% catalyst with additional water in 0.5 mL DCE: (■) yield, (▲) Ee. | ||
Evaluation of the substrate scope revealed that high levels of enantioselectivity could be achieved for a wide range of oxindole derivatives 1 (89–99% ee; Table 2) with the R2 group either linear alkyl (Table 2, entries 3, 4 and 6) or branched (Table 2, entry 5) at the C3 position, the corresponding adducts 3b–e were obtained in excellent results (91–97% ee, Table 2, entries 3–6). With 3-allyl or 3-propargyl substituents, the enantiomeric excess of the desired product was still excellent (Table 2, entries 7 and 8), which is meaningful since the unsaturated alkyl group is a useful handle for further functional group manipulation. To further evaluate the synthetic potential of the catalytic system, the reaction was conducted on gram-scale to afford 3a with similar results (Table 2, entry 2). In a previous report, the asymmetric hydroxymethylation of N-Boc-3-phenyloxindole was challenging, and the corresponding adduct was isolated in less than 5% ee, due to its high competing background reaction.3d Significantly, when 3-phenyloxindole 1h was tested by this method, 90% ee and 93% yield could be obtained in 6 h (Table 2, entry 9). For R2 being an aromatic ring, both electron-donating groups (Table 2, entries 11–15) and electron-withdrawing groups (Table 2, entries 16–19) on the ring afforded products in excellent 79–91% yield with 90–97% ee. Oxindoles 1s and 1t bearing bulky naphthylmethyl on C3 position reacted smoothly with formalin, giving the desired products with excellent enantioselectivities (up to 95% ee; Table 2, entries 20 and 21). Remarkably, for more challenging heterocyclic groups (1u and 1v), this catalyst system was also compatible and delivered the products in >99% ee (Table 2, entries 22 and 23). Electronically deficient 5-bromo-2-oxindole 1w was also tolerated (Table 2, entry 24).
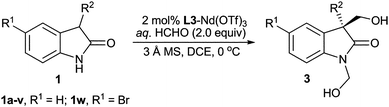
| Entrya | R2 | Product | t/h | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
|
a For details, see ESI1.
b Isolated yield.
c Determined by chiral HPLC analysis.
d Scale up to 10 mmol (1.47 g), catalyzed by 2 mol% L3-Nd(OTf)3.
e The reaction was carried out in 0.6 mL of mixed DCE and CHCl2CHCl2 (1:1, v/v).
|
|||||
| 1 | Me | 3a | 20 | 81 | 89 |
| 2 | Me | 3a | 20 | 78 | 87 |
| 3 | Et | 3b | 22 | 82 | 94 |
| 4 | n-Pr | 3c | 22 | 77 | 97 |
| 5 | i-Pr | 3d | 24 | 60 | 91 |
| 6 | n-Bu | 3e | 24 | 83 | 94 |
| 7 | Allyl | 3f | 24 | 87 | 93 |
| 8 | Propargyl | 3g | 24 | 67 | 96 |
| 9 | Ph | 3h | 6 | 91 | 90 |
| 10 | Bn | 3i | 24 | 85 | 95 |
| 11 | 2-MeC6H4CH2 | 3j | 24 | 79 | 95 |
| 12 | 4-MeOC6H4CH2 | 3k | 24 | 81 | 92 |
| 13e | Piperonyl | 3l | 36 | 87 | 92 |
| 14 | 3-PhOC6H4CH2 | 3m | 24 | 91 | 97 |
| 15 | 4-PhC6H4CH2 | 3n | 24 | 87 | 91 |
| 16 | 2-ClC6H4CH2 | 3o | 16 | 90 | 96 |
| 17 | 4-ClC6H4CH2 | 3p | 16 | 79 | 96 |
| 18 | 2,4-Cl2C6H3CH2 | 3q | 16 | 88 | 93 |
| 19 | 4-BrC6H4CH2 | 3r | 16 | 79 | 90 |
| 20e | 1-Naphthylmethyl | 3s | 16 | 92 | 92 |
| 21e | 2-Naphthylmethyl | 3t | 16 | 93 | 95 |
| 22e | 2-Pyridylmethyl | 3u | 36 | 75 | >99 |
| 23e | 2-Thienylmethyl | 3v | 24 | 89 | >99 |
| 24 | Me | 3w | 8 | 91 | 89 |
The rich chemistry of functional group interconvertion of the hydroxyl group promise products 3 as attractive targets for further application and development (Scheme 2). The N-hydroxymethyl group was easily removed by treating 3a with an aqueous methanol of NaOH to give 2a with no racemization.14d Meanwhile, the absolute configuration of 3a was determined to be S after converting 2a to known compound 6 with complete retention of stereochemistry over a four-step transformation.17 The configuration of the other oxindoles 3 was assigned tentatively by analogy. To further illustrate the synthetic utility of this methodology, we undertook the formal synthesis of enantioenriched physostigmine18 and coerulescine,19 which show wide biological activities such as in treatment of glaucoma and myasthenia gravis. By treating 3a with 1.2 equiv. of NaH prior to the addition of anhydrous LiBr and MeI, the N-methyl protected oxindole 7,18c as a key intermediate, was isolated in 70% yield with maintenance of ee (Scheme 2B). Notably, this protocol provided the lowest-step route to the enantioenriched product 7, compared with the reported methods.3d,18c The absolute configuration of 3a was also further confirmed by comparing the optical rotation of the synthesized compound 7 with the literature data. Similarly, the intermediate 819b of coerulescine was obtained in 75% yield with 93% ee (Scheme 2, C).
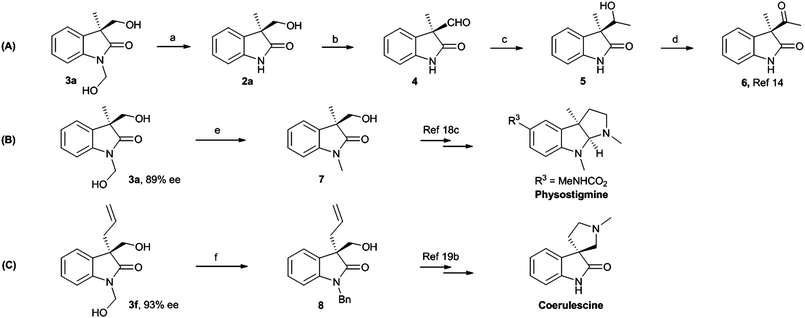 | ||
| Scheme 2 Synthetic application of the products and determination of the absolute configuration. Reagents and conditions: (a) NaOH aq., MeOH, rt, 2a: 85% yield, 89% ee; (b) Dess–Martin, CH2Cl2, 0–25 °C, 4: 60% yield; (c) MeMgBr (3 M), THF, −78 °C, 5: 85% yield; (d) Dess–Martin, CH2Cl2, 0–25 °C, 6: 95% yield, 89% ee; (e) (i) NaH, THF, 0–25 °C; (ii) LiBr, MeI, THF, rt; 7: 70% yield, 89% ee; (f) (i) NaH, THF, 0–25 °C; (ii) LiBr, BnBr, THF, rt; 8: 75% yield, 93% ee. | ||
Control experiments were performed to elucidate the reaction process. When the amount of formalin was reduced to 0.8 equiv, 2a was isolated in 5% yield with 75% ee (Scheme 3A). In this case, the major product was still 3a with 75% ee. Upon treatment with either 5 mol% 1,1,3,3-tetramethylguanidine or L3-Nd(OTf)3, 2a could be converted to 3a immediately with the maintenance of ee. This implies that C-addition occurs predominantly at the C3 position of oxindole; no N-adduct was observed during the reaction. This phenomenon revealed that the reaction proceeded through C-addition and N-addition sequentially. Then the amount of formalin was investigated from a range of 0.8 to 3.0 equivalents. The reactivity increased upon increasing the amount of formaldehyde and the optimal amount of formalin was 2.0 equivalents with regard to both yield and enantiomeric excess.
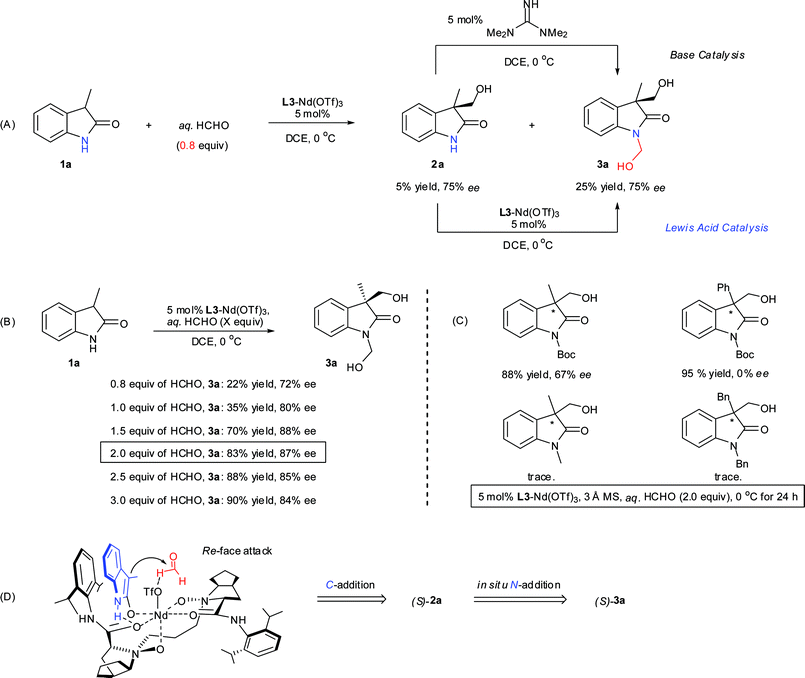 | ||
| Scheme 3 (A) Transformation of 2a to 3a. (B) Effect of the amount of formalin. (C) Investigation of N-protected 2-oxindoles. (D) Proposed transition state for the catalytic asymmetric hydroxymethylation reaction of unprotected oxindole 1a. | ||
To gain preliminary insight into the reaction mechanism, N-protected-3-substituted-2-oxindoles were investigated under the optimized conditions (Scheme 3C). To our surprise, low ee or even no adduct was obtained. Typically, in the case of N-Boc-3-phenyl-2-oxindole, the corresponding adduct was isolated in high yield but as a racemic mixture, which might be ascribed to the high competing background reaction. Other N-protected substrates such as N-methyl and N-Bn protected ones completely prevented the reaction to occur. This fact indicated the important role of the NH moiety of oxindole for this reaction.
For direct proof of the mechanism of the proposed NdIII-based enolate activation, we investigated the hydroxymethylation reaction using ESIMS, which enables identification of the critical intermediates in the reaction mixture.20 Initially, the catalyst was prepared from Nd(OTf)3 and L3 in a 1:1 ratio in DCE. The spectrum of the sample obtained from the reaction mixture after stirring for 1 h revealed the parent ion at m/z 1140.3007, which corresponds to that of the catalyst [L3 + Nd(OTf)2]+. Moreover, in the presence of 1a (2.0 equiv. relative to the catalyst) and formaldehyde (4.0 equiv. relative to the catalyst) in DCE, characteristic signals at m/z 1168.4681 and 1198.4736 were observed in the ESIMS spectrum of ions [L3 + NdOTf + 1a + CH2O]+ and [L3 + NdOTf + 1a + 2CH2O]+. These signals were consistent with the fragmentation of the hydroxymethylation adducts before scission with the L3-NdIII complex, which indicated that these signals should correspond to intermediates of the adducts. The above MS results confirmed our proposed activation models as shown in Scheme 1.
On the basis of the experimental results and the analogy of the previous ScIII-N,N′-dioxide complex,9e the possible transition state is proposed in Scheme 3D. Both the carbonyl oxygen and N-oxide oxygen are expected to coordinate with neodymium(III) in the complex. Oxindole 1a should coordinate to the neodymium(III) though oxygen to form a chiral enolate. The Re face of 1a is shielded by the neighboring 2,6-diisopropylphenyl group of the ligand, and the NH moiety of 1a and interacts with L3 through hydrogen bonding. Nucleophilic attack from the Re face predominantly would give the S-configured adduct, which is in accordance with the experimental results. Then 2a undergoes quick N-hydroxymethylation to form the final product 3a in the presence of L3-Nd(OTf)3 and excess formaldehyde.
Experimental
Representative experimental procedure (Table 1, entry 15)
A mixture of L3 (5 mol%), Nd(OTf)3 (5 mol%), 3 Å molecular sieves (3 mg) and oxindole 1a (0.1 mmol) was stirred in DCE (0.6 mL) at 35 °C for 1 h. Formalin (2.0 equiv.) was added at 0 °C. The reaction was stirred at 0 °C for 20 h. The crude product was directly purified by flash chromatography (petroleum ether–ethyl acetate, 1:2) to afford product 3a in 80% yield with 89% ee.
Catalyst-recovery procedure (Table 1, entry 13)
A gram-scale reaction involving 1a (10 mmol, 1.47 g) and formalin (2.0 equiv.) was promoted by 2 mol% L3-Nd(OTf)3 in 30 mL DCE at 0 °C for 36 h. Then pure water (10 mL) was added. The aqueous layer was extracted with CH2Cl2 (10 mL × 2), and Nd(OTf)3 was recovered from aqueous phase after removing water. The combined organic phase was concentrated and followed by chromatography on silica gel (petroleum ether–ethyl acetate, 1:2) to give product 3a, and then (methanol–ethyl acetate, 1:10) to give recovered L3.
Conclusion
The general and highly enantioselective hydroxymethylation reaction of unprotected oxindoles was successfully established. The significant feature of this approach is the use of cheap and readily available formalin as a hydroxymethylation C1 unit. The unprotected 3-substituted-2-oxindole was activated by a NdIII-N,N′-dioxide complex through an O-bound enolate. In the presence of 2 mol% L3-Nd(OTf)3 complex, 1,3-bis(hydroxymethyl)-2-oxindoles were obtained in up to 97% yield with >99% ee under mild conditions. The preliminary investigation revealed that the reaction was carried out through C-addition and N-addition sequentially. In view of the rich chemistry of functional group interconversion of the hydroxy group, this reaction might provide a new practical and broad applicable access to chiral linchpins bearing oxindoles. Moreover, the unexpected N-hydroxymethylated moiety of the adducts might promise valuable expansion to the N-hydroxymethyl derivatives of nitrogen heterocycles as possible prodrugs. Further studies of the reaction mechanism and application of this catalyst to other reactions are underway.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (No. 20732003), PCSIRT (No. IRT0846) and the National Basic Research Program of China (973 Program: No. 2010CB833300). We also thank Sichuan University Analytical & Testing Centre for NMR analysis, and the State Key Laboratory of biotherapy for HRMS analysis.Notes and references
- For reviews, see: (a) J. L. G. Fierro, Catal. Lett., 1993, 22, 67 CAS; (b) J. S. Lee, J. C. Kim and Y. G. Kim, Appl. Catal., 1990, 57, 1 Search PubMed.
- (a) N. Ozawa, M. Wadamoto, K. Ishihara and H. Yamamoto, Synlett, 2003, 2219 CAS; (b) S. Ishikawa, T. Hamada, K. Manabe and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 12236 CrossRef CAS; (c) S. Kobayashi, T. Ogino, H. Shimizu, S. Ishikawa, T. Hamada and K. Manabe, Org. Lett., 2005, 7, 4729 CrossRef CAS; (d) M. Kokubo, C. Ogawa and S. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 6909 CrossRef CAS.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS; (b) J. Casas, H. Sundén and A. Córdova, Tetrahedron Lett., 2004, 45, 6117 CrossRef CAS; (c) S. Kobayashi, M. Kokubo, K. Kawasumi and T. Nagano, Chem.–Asian J., 2010, 5, 490 CrossRef CAS; (d) X. L. Liu, Y. H. Liao, Z. J. Wu, L. F. Cun, X. M. Zhang and W. C. Yuan, J. Org. Chem., 2010, 75, 4872 CrossRef CAS.
- R. Kuwano, H. Miyazaki and Y. Ito, Chem. Commun., 1998, 71 RSC.
- (a) I. Fukuchi, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2007, 349, 509 CrossRef CAS; (b) S. Mouri, Z. H. Chen, S. Matsunaga and M. Shibasaki, Chem. Commun., 2009, 5138 RSC.
- For selected reviews and examples, see: (a) R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127 CrossRef CAS; (b) A. K. Franz, P. D. Dreyfuss and S. L. Schreiber, J. Am. Chem. Soc., 2007, 129, 1020 CrossRef CAS; (c) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323 CrossRef CAS; (d) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CAS.
- The unprotected 3-substituted-2-oxindoles was less acidic carbon nucleophiles than N-Boc-3-substituted-2-oxindoles; namely, N-Boc-3-substituted-2-oxindoles have higher reactivities; see ref. 6d.
- (a) S. Kobayashi, Lanthanide triflate-catalyzed carbon–carbon bond-forming reactions in organic synthesis, in Lanthanides: Chemistry and Use in Organic Synthesis, ed. S. Kobayashi, Springer, Heidelberg, 1999, p. 63 Search PubMed; (b) K. Mikami, M. Terada and H. Matsuzawa, Angew. Chem., Int. Ed., 2002, 41, 3554 CrossRef CAS.
- For selected examples of our REIII-N,N′-dioxide complex studies, see: (a) Z. P. Yu, X. H. Liu, Z. H. Dong, M. S. Xie and X. M. Feng, Angew. Chem., Int. Ed., 2008, 47, 1308 CrossRef CAS; (b) X. Yang, X. Zhou, L. L. Lin, L. Chang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2008, 47, 7079 CrossRef CAS; (c) M. S. Xie, X. H. Chen, Y. Zhu, B. Gao, L. L. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 3799 CrossRef CAS; (d) Y. H. Hui, J. Jiang, W. T. Wang, W. L. Chen, Y. F. Cai, L. L. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 4290 CrossRef CAS; (e) Y. L. Liu, D. J. Shang, X. Zhou, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2009, 15, 2055 CrossRef CAS.
- For in situ formed metal enolate, see: (a) S. Saito and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 8704 CrossRef CAS; (b) A. Yamaguchi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 10842 CrossRef CAS; (c) T. Yoshino, H. Morimoto, G. Lu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 17082 CrossRef CAS.
- R. Kapiller-Dezsőfi and B. Volk, New J. Chem., 2004, 28, 1214 RSC.
- For a review of direct aldol reactions, see: Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004. For recent examples of direct aldol reaction of 3-substituted-2-oxindoles, see: Search PubMed (a) S. Ogawa, N. Shibata, J. Inagaki, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2007, 46, 8666 CrossRef CAS; (b) K. Shen, X. H. Liu, K. Zheng, W. Li, X. L. Hu, L. L. Lin and X. M Feng, Chem.–Eur. J., 2010, 16, 3736 CrossRef CAS . Also see refs. 3c and 3d.
- In the reaction system, the ee of 3a did not change over time; see ESI†.
- For selected examples, see: (a) J. D. Milkowski, D. F. Veber and R. Hirschmann, Org. Synth., 1979, 59, 190; (b) B. Jouglet and G. Rousseau, Tetrahedron Lett., 1993, 34, 2307 CrossRef CAS; (c) B. Jouglet, S. Oumoch and G. Rousseau, Synth. Commun., 1995, 25, 3869 CrossRef CAS; (d) S. I. Hyun and Y. G. Kim, Tetrahedron Lett., 1998, 39, 4299 CrossRef CAS.
- P. C. Bansal, I. H. Pitman, J. N. S. Tam, M. Mertes and J. J. Kaminski, J. Pharm. Sci., 1981, 70, 850 CAS.
- When 1.0 equivalent CH3(CH2)11SO3Na or tetrabutylammonium chloride was introduced to the reaction, the ee of 3a decreased to 20 and 15%, respectively.
- T. A. Duffey, S. A. Shaw and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14 CrossRef CAS.
- (a) J. Jobst and O. Hesse, Justus Liebigs Ann. Chem., 1864, 129, 115 CrossRef; (b) E. Stedman and G. Barger, J. Chem. Soc., 1925, 127, 247 RSC; (c) K. Asakawa, N. Noguchi and M. Nakada, Heterocycles, 2008, 76, 183 CrossRef CAS.
- (a) A. Jossang, P. Jossang, H. A. Hadi, T. Sévenet and B. Bodo, J. Org. Chem., 1991, 56, 6527 CrossRef CAS; (b) J. E. Thomson, A. F. Kyle, K. B. Ling, S. R. Smith, A. M. Z. Slawin and A. D. Smith, Tetrahedron, 2010, 66, 3801 CrossRef CAS.
- For ESIMS spectra, see ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and spectroscopic data. See DOI: 10.1039/c0sc00385a |
| This journal is © The Royal Society of Chemistry 2010 |