Gold(I)-catalyzed formation of dihydroquinolines and indoles from N-aminophenyl propargyl malonates†‡
Colombe
Gronnier
,
Yann
Odabachian
and
Fabien
Gagosz
*
Laboratoire de Synthèse Organique, UMR 7652 CNRS/Ecole Polytechnique, Ecole Polytechnique, 91128 Palaiseau, France. E-mail: gagosz@dcso.polytechnique.fr
First published on 21st April 2010
Abstract
The use of [XPhosAu(NCMe)]SbF6 in nitromethane at 100 °C allows the rapid and efficient formation of variously substituted dihydroquinolines, which can be subsequently converted into indoles by a rare photochemical rearrangement.
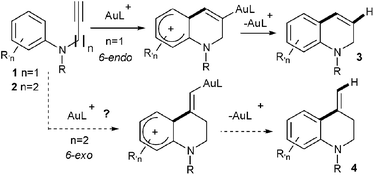
Gold catalysis has proven during the last decade to be a powerful synthetic tool for the synthesis of a large variety of nitrogen containing heterocycles.1 Among the different strategies employed, those based on the use of an intramolecular gold-catalyzed hydroarylation reaction are particularly attractive as they allow the easy creation of a new C–C bond by the nucleophilic addition of an aryl unit onto an insaturation.2 This approach has been applied for instance to the formation of dihydroquinolines 3 by a gold-catalyzed 6-endocyclization of N-propargyl anilines 1 (Scheme 1, n = 1).3
 | ||
| Scheme 1 Au-mediated synthetic approaches to hydroquinolines. | ||
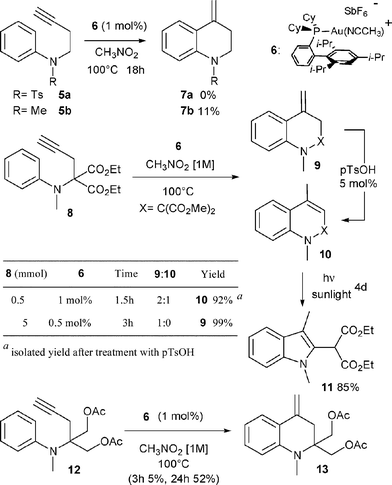
Following our own interest in gold catalysis, we wondered if the homologous N-butynyl anilines 2 might be valuable substrates for the synthesis of the isomeric exo-methylene tetrahydroquinolines 4via, in this case, a 6-exo cyclization (Scheme 1, n = 2). N-Butynyl tosylaniline 5a, was first chosen as a model substrate for this study (Scheme 2).3a,b However, no cyclized product 7a could be observed in this case whatever the catalyst, the solvent or the temperature used.4 The transformation was next attempted with substrate 5b where the aryl moiety is more nucleophilic. An encouraging 11% yield of 7b was obtained when the reaction was conducted in nitromethane at 100 °C with [XPhosAu(NCMe)]SbF6 (6) as the catalyst.5Aniline derivative 8, proved to be a more useful substrate since its treatment with 1 mol% of 6 in nitromethane at 100 °C for 1.5 h led to the formation of two cyclized products namely exo-methylene tetrahydroquinoline 9 and dihydroquinoline 10 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 2).6,7
:1 ratio (Scheme 2).6,7
 | ||
| Scheme 2 Preliminary studies. | ||
Subsequent treatment of the crude reaction mixture with p=TsOH (5 mol%) led to the rapid isomerization of 9 into 10, which was finally isolated in 92% yield.8 The reaction could also be efficiently performed (99%) on a 5 mmol scale with a reduced loading of catalyst (0.5 mol%). Under these conditions and by carefully monitoring the reaction, it was even possible to suppress the formation of isomeric compound 10. The presence of the malonate moiety in substrate 8 appears to be crucial for the efficient formation of the hydroarylation product. The replacement of the esters of the malonate moiety by two acetoxymethyl groups (substrate 12) had a negative effect, as only 5% of the corresponding hydroarylation product was produced after 3 h of reaction.9Dihydroquinoline 10 was not very stable, and we were surprised to observe its slow conversion into indole 11 when a chloroform solution of 10 was exposed to sunlight (Scheme 2).10,11
This unexpected photochemical rearrangement is particularly interesting since indole 11 could be formed in high yield in only two steps from the readily available N-aminophenyl propargyl malonate 8.
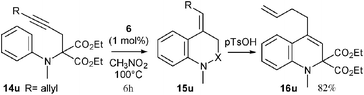
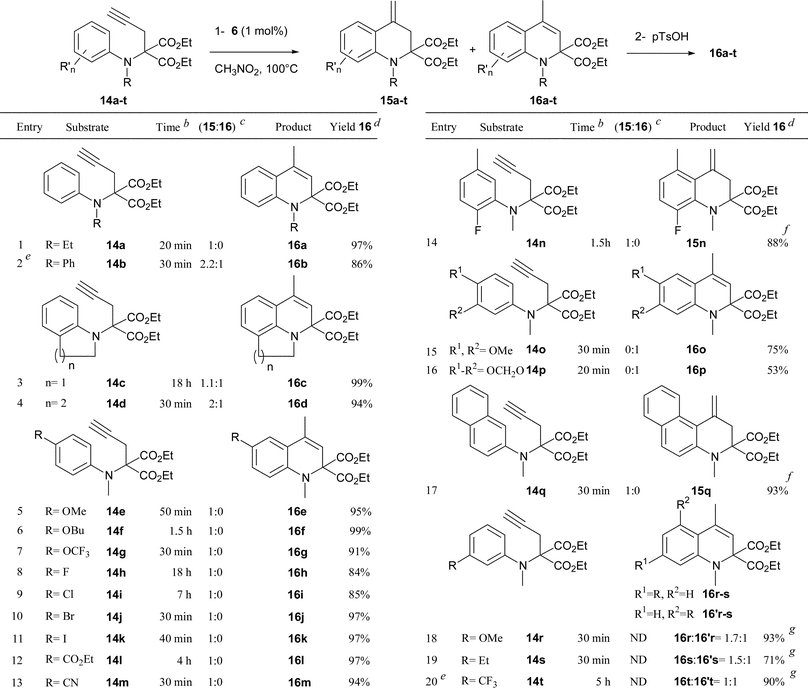
A series of diversely substituted N-aminophenyl propargyl malonates 14a–t were then synthesized4 and reacted under the same reaction conditions (1 mol% of 6 in nitromethane at 100 °C) to determine the scope of the hydroarylation process. The results of this study are compiled in Table 1. The transformation proved to be efficient and various dihydroquinolines 16a–t were rapidly obtained in yields ranging from 53 to 99%. The gold catalyzed hydroarylation process selectively furnished the exo-methylene compounds 15 with the exception of substrates 14b–d, which resulted in isomeric mixtures of 15b–d and 16b–d (entries 2–4). In the case of the electron rich substrates 14o and 14p (entries 15 and 16), the gold catalyzed process directly produced the corresponding dihydroquinolines 16o and 16p, as the result of a more favourable gold-catalyzed isomerization of the exo-methylene. Various substitutents on the nitrogen atom such as an alkyl group (entry 1), a phenyl ring (entry 2) or a cycloalkyl unit linked to the aromatic ring (entries 3 and 4) were tolerated. The reaction was also compatible with a wide range of functional groups onto the aromatic ring such as an electron donating ether group (entries 5–7, 15, 16 and 18), a halogen atom (entries 8–11 and 14), or an electron withdrawing ester (entry 12), cyano (entry 13) or trifluoromethyl (entry 20) group. Notably, the efficiency and the rapidity of the reaction were not markedly dependent on the electronic nature of the substituent (compare entries 5–13) or its position on the aromatic ring since ortho (entries 3, 4 and 14), meta (entries 15–20) or para (entries 5–13) substituted aryl substrates reacted equally well. While compounds 14o–q, possessing two substituents at the meta and para positions of the aniline moiety only produced dihydroquinolines 16o–q (entries 15–17), the reaction proved to be completely unselective in the case of substrates 14r–t (entries 18–20).12 The transformation could also be applied with the same efficiency to substituted alkyne 14u (eqn (1)). The selectivity of the reaction was complete as the result of an anti addition of the nucleophilic aryl moiety onto the gold-activated alkyne.
 | (1) |
| a Reaction conditions: step 1: 14 (1 equiv.), [XPhosAu(NCMe)]SbF6 (6) (0.01 equiv.) in refluxing CH3NO2 (1 M); step 2: p-TsOH (0.05 equiv.) in CH2Cl2 (0.1 M) at rt or reflux. b Reaction time for step 1. c Determined by 1H NMR spectroscopy of the crude reaction mixture after step 1. d Isolated yields after step 2. e 6 (2 mol%). f Yield of exo-methylene compound 15. Isomerization was incomplete. g Global yield of the isomeric mixture (16 + 16'). |
|---|
|
We finally turned our attention to the rearrangement of the dihydroquinolines into the corresponding indoles, as initially observed for compound 10. A series of dihydroquinolines were indeed similarly converted into the corresponding indoles 20a–d by simple exposure to sunlight (67–88%). A mechanism for this remarkable ring contraction is proposed in Scheme 3. Upon irradiation, dihydroquinoline 17 undergoes presumably an electrocyclic ring opening leading to intermediate 18. A subsequent nucleophilic attack of the nitrogen atom onto the electrophilic alkylidene malonate moiety, energetically driven by the re-aromatisation of the benzene ring, furnishes a zwitterionic species 19 which finally collapses into indole 20.13
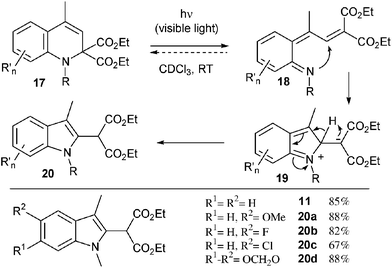 | ||
| Scheme 3 Photochemical rearrangement to indoles. | ||
In summary, we have developed an efficient synthesis of exo-methylene tetrahydroquinolines and dihydroquinolines from readily accessible N-aminophenyl propargyl malonates. The hydroarylation process, which could be performed with a low loading of catalyst (1 mol%), proved to be rapid and general allowing the presence of a plethora of functional groups on the aromatic ring. Furthermore, the dihydroquinolines were shown to easily rearrange to functionalized indoles under photochemical conditions thus expanding the general synthetic utility of the approach.
This work was supported by the CNRS and Ecole Polytechnique. The authors thank Prof. S. Z. Zard for helpful discussions.
Notes and references
- Recent reviews: (a) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (b) P. Belmont and E. Parker, Eur. J. Org. Chem., 2009, 6075 CrossRef CAS; (c) V. Michelet, P. Y. Toullec and J. P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS; (d) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (e) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef CAS; (f) Z. Li, C. Brower and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS; (g) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS; (h) D. J. Gorin and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS; (i) R. Skouta and C.-J. Li, Tetrahedron, 2008, 64, 4917 CrossRef CAS.
- Selected articles: (a) M. A. Tarselli and M. R. Gagne, J. Org. Chem., 2008, 73, 2439 CrossRef CAS; (b) I. V. Seregin, A. W. Schammel and V. Gevorgyan, Org. Lett., 2007, 9, 3433 CrossRef CAS; (c) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2007, 9, 4821 CrossRef CAS; (d) N. Marion, S. Díez-González, P. de Frémont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647 CrossRef CAS; (e) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105 CrossRef CAS; (f) Z. Liu, A. S. Wasmuth and S. G. Nelson, J. Am. Chem. Soc., 2006, 128, 10352 CrossRef CAS; (g) D. J. Gorin, P. Dube and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 14480 CrossRef CAS; (h) Z. Zhang, C. Liu, R. E. Kinder, X. Han, H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2006, 128, 9066 CrossRef CAS; (i) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556 CrossRef; (j) Z. Shi and C. He, J. Org. Chem., 2004, 69, 3669 CrossRef CAS.
- Selected articles: (a) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS; (b) C. Nevado and A. M. Echavarren, Chem.–Eur. J., 2005, 11, 3155 CrossRef CAS; (c) R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901 CrossRef CAS; (d) F. Xiao, Y. Chen, Y. Liu and J. Wang, Tetrahedron, 2008, 64, 2755 CrossRef CAS; (e) X.-Y. Liu, P. Ding, J.-S. Huang and C.-M. Che, Org. Lett., 2007, 9, 2645 CrossRef CAS. See also; with Ag catalysis: (f) Y. Luo, Z. Li and C.-J. Li, Org. Lett., 2005, 7, 2675 CrossRef CAS; with Fe catalysis: (g) K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748 RSC; with Lewis-acid catalysis: (h) T. Ishikawa, S. Manabe, T. Aikawa, T. Kudo and S. Saito, Org. Lett., 2004, 6, 2361 CrossRef CAS.
- See ESI‡ for more details.
- For the use of catalyst 6 in CH3NO2, see: I. D. Jurberg, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2010, 132, 3543 Search PubMed.
- The use of catalyst 6 in CH3NO2 appears to be the catalytic system of choice as it stabilizes the catalyst and leads to a rapid and generally selective formation of 15 for the series of substrates studied.
- The 9:10 ratio slowly evolves upon prolonged reaction time. For this alkene isomerisation, see: A. S. K. Hashmi, M. C. Blanco, E. Kurpejovic, W. Frey and J. W. Bats, Adv. Synth. Catal., 2006, 348, 709 Search PubMed.
- Dihydroquinoline 10 was unstable in the presence of silica. A simple basic work-up followed, if necessary, by a rapid filtration through a pad of silica was used to obtain the product in pure form.
- The malonate moiety plays multiple roles in this transformation: it induces a Thorpe–Ingold effect resulting in a more favorable cyclization while limiting the coordination of the gold catalyst with the nitrogen atom probably through steric and electronic effects.
- The electrocyclic ring-opening of 1,2-dihydroquinolines is a rare process, see: (a) M. Ikeda, S. Matsugashita, H. Ishibashi and Y. Tamura, J. Chem. Soc., Chem. Commun., 1973, 922 RSC; (b) M. Ikeda, S. Matsugashita, F. Tabusa, H. Ishibashi and Y. Tamura, J. Chem. Soc., Chem. Commun., 1974, 433 RSC; (c) M. Ikeda, S. Matsugashita, F. Tabusa and Y. Tamura, J. Chem. Soc., Perkin Trans. 1, 1977, 1166 RSC.
- No reaction took place under thermal conditions (toluene, 110 °C) or in the presence of a Lewis acid (Yb(OTf)3, rt). Compound 10 was stable in the absence of light.
- The hydroarylation seems to be little influenced by the electronic nature or the steric hindrance of a meta substituent.
- Formation of 20 may also be explained by the intermediate formation of an allenylidene malonate from 18 (see ref. 10).
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and spectra. See DOI: 10.1039/c0cc00033g |
| This journal is © The Royal Society of Chemistry 2011 |