Accessing the antipodal series in microbial arene oxidation: a novel diene rearrangement induced by tricarbonyliron(0) complexation†‡
Monika Ali
Khan
a,
John P.
Lowe
a,
Andrew L.
Johnson
a,
Alan J. W.
Stewart
b and
Simon E.
Lewis
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: S.E.Lewis@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386568
bProsidion Limited, Windrush Court, Watlington Road, Oxford, OX4 6LT, UK
First published on 20th August 2010
Abstract
A cyclohexadiene ligand prepared by microbial arene 1,2-dihydroxylation undergoes spontaneous rearrangement upon complexation to tricarbonyliron(0). Subsequent iron removal affords a novel route to formal arene 2,3-dihydroxylation products enantiomeric to those obtainable by direct microbial arene oxidation.
Since the first report in 1968,1 enzymatic dihydroxylation of aromatic substrates to afford enantiopure building blocks for synthesis has become an established methodology.2 In excess of 400 arene cis-diol products have been reported. The vast majority of these are produced by organisms expressing toluene dioxygenase (TDO), naphthalene dioxygenase (NDO) and biphenyl dioxygenase (BPDO) enzymes. These metabolise substituted arene substrates in a regio- and stereoselective fashion. A reliable predictive model has been reported for such transformations3 and the sense of enantioinduction is conserved across organisms and substrates (Scheme 1a). In contrast, organisms expressing benzoate dioxygenase (BZDO) enzymes oxidise benzoic acids in a process that exhibits not only different regioselectivity, but also the opposite sense of enantioinduction. For example, R. eutrophus B9,4P. putidaU1035 and P. putidaKTSY01 (pSYM01)6 oxidise benzoic acid to benzoate 1,2-cis-dihydrodiol 4 (Scheme 1b). Diol 4 has proved to be a versatile chiron for synthesis, having seen several applications,7 most notably in the synthesis of tetracycline antibiotics.8
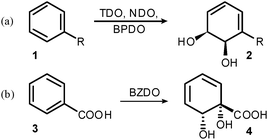 | ||
| Scheme 1 Regio- and stereoselectivity of dioxygenases. | ||
Dienes such as 2 and 4 may be derivatised as the corresponding tricarbonyliron(0) complexes9 and we have recently shown that the methyl ester of 4 coordinates to iron with complete facial selectivity.10 The sole isomer obtained is that in which the diol is endo (6, Scheme 2a), which is noteworthy since the diene presents Lewis basic functionality on both faces. Coordination to iron permits stereoselective ligand modification at the carbons adjacent to the diene, by means of cationic η5-dienyl intermediates.11 In this context we sought to access a complex in which the diol was exo, by protecting the diol as an acetonide (Scheme 2b). We reasoned that additional steric bulk on the lower face would disfavour the precoordination of the iron to a Lewis basic diol oxygen lone pair, which has been proposed to rationalise the facial selectivity observed previously.
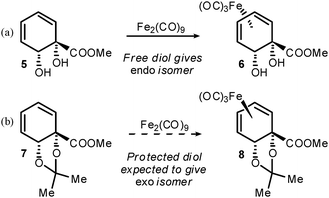 | ||
| Scheme 2 Lewis basic functionality directs iron coordination. | ||
In the event, treatment of acetonide 7 with Fe2(CO)9 in THF gave 9, in which the acetonide was indeed exo, but an isomerisation had occurred such that the ester was now conjugated to the diene (Scheme 3). The structure of 9 was determined by X-ray crystallography (Fig. 1).12
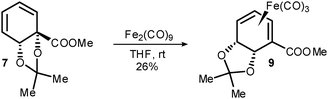 | ||
| Scheme 3 Diene rearrangement upon complexation. | ||
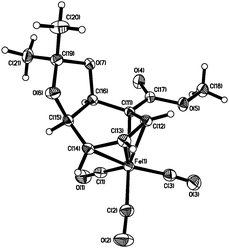 | ||
| Fig. 1 X-Ray structure of 9. (50% Thermal ellipsoids for all non-hydrogen atoms.) | ||
Isomerised complex 9 underwent facile oxidative demetallation with trimethylamine N-oxide to afford uncomplexed diene (2S,3R)-11 (Scheme 4). The enantiopurity of (2S,3R)-11 was determined to be >95% ee by means of ester reduction and subsequent formation of Mosher's ester derivatives.‡ Attempted removal of the acetonide in (2S,3R)-11 upon exposure to Brønsted acid proved unsuccessful due to facile dehydration/rearomatisation. However, in preliminary experiments, iodine in methanol13 has been observed by NMR to afford 12 from (2S,3R)-11, albeit with a degree of concomitant rearomatisation.
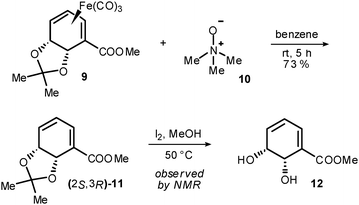 | ||
| Scheme 4 Deprotection of 9. | ||
Direct microbial oxidation of methyl benzoate to afford a 2,3-diol and subsequent acetonide formation has been reported.14,15 However, in this instance, the opposite enantiomer, (2R,3S)-11, was obtained. Indeed, the enantiomer reported here, (2S,3R)-11, has not been described to date; this iron-mediated diene rearrangement represents a new route to an arene 2,3-cis-diol derivative antipodal to that obtained by direct biooxidation. Thus far, the synthetic utility of arene 2,3-cis-diols has been constrained by the comparative difficulty in accessing the non-natural enantiomeric series. We anticipate that the transformation reported here will be of great synthetic utility, for example in allowing the synthesis of D-configured carbasugars (many L-carbasugars have been synthesised from diols of type 2; the synthesis of (2S,3R)-11 reported here constitutes formal syntheses of carba-β-D-galactopyranose, carba-β-D-talopyranose and carba-α-D-talopyranose).15 In addition, elaboration of the ester will permit access to numerous antipodal arene 2,3-cis-diols not accessible by other means.
As regards the mechanism of formation of 9, isotopic labelling studies were undertaken to ascertain the identity of the migrating group. Biooxidation of p-deuterobenzoic afforded 13, which then permitted the preparation of 15 (Scheme 5). Regioisomer 14 was not formed, confirming that 9 arises through migration of the acetonide and not the carboxymethyl group.
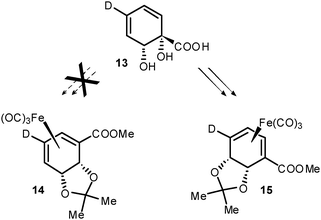 | ||
| Scheme 5 Isotopic labelling of substrate confirms acetonide migration. | ||
In view of the above, a mechanism may be proposed that invokes a cationic η5-cyclohexadienyl intermediate,1117. This could plausibly arise from initial formation of the expected 1,2-isomer 8, subsequent co-ordination of an unspecified Lewis-acidic species to an acetonide oxygen and C–O bond scission. Attack of the tethered nucleophile (exo to iron and ω- to the electron-withdrawing group, as is precedented)16 would then generate 9 (Scheme 5). In addition to 17, the regioisomeric η5 cation 19 may also be formed as a transient intermediate by means of C–O bond scission at the other acetonide oxygen. The two cations 17 and 19 differ appreciably in electronic structure as the ester is conjugated to the dienyl system only in 17. Crucially, 19 is achiral; recombination of the tethered nucleophile at either terminus of the η5dienyl ligand in 19 will effect racemisation of 8.17 That (2S,3R)-11 and, by inference, 9 are not in fact racemic is suggestive of the reaction being under kinetic control. Specifically, formation of 17 may be kinetically favoured over 19 due to relief of steric strain as the ester α-carbon rehybridises such that the ester is in the plane of the dienyl system. If formation of 17 from 16 is effectively irreversible (due to the aforementioned preference for nucleophile recombination ω- to the ester) and k1 ≫ k2 (Scheme 6), the ee of 9 will not be eroded.
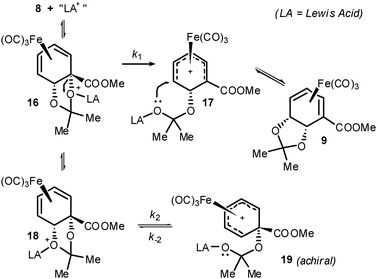 | ||
| Scheme 6 Possible formation of 9via cationic η5-dienyl intermediate. | ||
Variable temperature 13C{1H} NMR spectra for the carbonyl region of complex 9 clearly show fluxional behaviour. This fluxionality may be ascribed to turnstile rotation of the iron carbonyl ligands. The variable temperature spectra of 9 were simulated‡ in order to derive the activation parameters for the exchange process of Ea = 46.8 ± 1.7 kJ mol−1, ΔH‡ = 44.4 ± 1.6 kJ mol−l, ΔS‡ = −22.8 ± 5.2 J K−1 mol−1 and ΔG‡(298) = 51.2 ± 3.1 kJ mol−1. Use of 13C{1H} VT-NMR to probe fluxionality in tricarbonyliron(0) complexes is well established and thermodynamic parameters have been derived by this method for numerous other tricarbonyliron(0)diene complexes.18,19 The calculated value of ΔG‡(298) for 9 is comparable to that for the analogous tropone complex (ΔG‡(298) = 53.1 ± 2.1 kJ mol−1).18
In summary, we have defined a rearrangement route to arene 2,3-cis-diol derivatives of non-natural configuration. The complex through which this rearrangement is realised exhibits hindered ligand rotation, for which thermodynamic data are presented. Our approach is complementary to other strategies reported previously for achieving this “enantiomeric switch”. For example, substituted iodobenzenes can undergo 2,3-dihydroxylation followed by reductive iodine removal,20,22 but this can preclude the use of diol derivatives possessing reductively labile functionality. The conceptually distinct approach of enantiodivergent synthesis has also been employed,21,22 requiring that two different synthetic routes be established. In contrast, the approach we describe utilises only oxidative conditions and will permit access to both enantiomers of a given target by the same synthetic pathway. Investigations to elucidate further the mechanism of formation of 9 and to demonstrate the scope of this transformation are underway in our laboratory and will be reported in due course.
We thank Prosidion Limited, EPSRC and CIKTN for a CASE studentship (to MAK). We also thank Prof. Andrew G. Myers (Harvard) for a generous gift of R. eutrophus B9 cells.
Notes and references
- D. T. Gibson, J. R. Koch, C. L. Schuld and R. E. Kallio, Biochemistry, 1968, 7, 3795 CrossRef CAS.
- For reviews, see: T. Hudlický and J. W. Reed, Synlett, 2009, 685 Search PubMed; D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181 CrossRef CAS; R. A. Johnson, Org. React., 2004, 63, 117 RSC; T. Hudlický, D. Gonzales and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 CAS.
- D. R. Boyd, N. D. Sharma, M. V. Hand, M. R. Groocock, N. A. Kerley, H. Dalton, J. Chima and G. N. Sheldrake, J. Chem. Soc., Chem. Commun., 1993, 974 RSC.
- A. M. Reiner and G. D. Hegeman, Biochemistry, 1971, 10, 2530 CrossRef CAS.
- J. T. Rossiter, S. R. Williams, A. E. G. Cass and D. W. Ribbons, Tetrahedron Lett., 1987, 28, 5173 CrossRef CAS.
- S.-Y. Sun, X. Zhang, Q. Zhou, J.-C. Chen and G.-Q. Chen, Appl. Microbiol. Biotechnol., 2008, 80, 977 CrossRef CAS.
- G. N. Jenkins, D. W. Ribbons, D. A. Widdowson, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1995, 2647 RSC; A. G. Myers, D. R. Siegel, D. J. Buzard and M. G. Charest, Org. Lett., 2001, 3, 2923 CrossRef CAS; M. H. Parker, B. E. Maryanoff and A. B. Reitz, Synlett, 2004, 2095 CrossRef CAS; M. D. Mihovilovic, H. G. Leisch and K. Mereiter, Tetrahedron Lett., 2004, 45, 7087 CrossRef CAS; T. C. M. Fischer, H. G. Leisch and M. D. Mihovilovic, Monatsh. Chem., 2010, 141, 699 CrossRef CAS.
- M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395 CrossRef CAS; M. G. Charest, D. R. Siegel and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 8292 CrossRef CAS.
- P. W. Howard, G. R. Stephenson and S. C. Taylor, J. Chem. Soc., Chem. Commun., 1988, 1603 RSC; P. W. Howard, G. R. Stephenson and S. C. Taylor, J. Organomet. Chem., 1989, 370, 97 CrossRef CAS; G. R. Stephenson, P. W. Howard and S. C. Taylor, J. Organomet. Chem., 1991, 419, C14 CrossRef CAS; A. J. Pearson, A. M. Gelormini and A. A. Pinkerton, Organometallics, 1992, 11, 936 CrossRef CAS.
- M. Ali Khan, M. F. Mahon, A. J. W. Stewart and S. E. Lewis, Organometallics, 2010, 29, 199 CrossRef CAS.
- For reviews, see: W. A. Donaldson and S. Chaudhury, Eur. J. Org. Chem., 2009, 3831 Search PubMed; I. Bauer and H.-J. Knölker, in Iron Catalysis in Organic Chemistry, ed. B. Plietker, Wiley-VCH, Weinheim, Germany, 2008, pp. 1–27 CrossRef CAS; R. Grée and J. P. Lellouche, in Advances in Metal-Organic Chemistry, ed. L. S. Liebskind, JAI Press, Greenwich, CT, 1995, pp. 129–273 CrossRef CAS; A. J. Pearson, Synlett, 1990, 10 CrossRef CAS.
- Crystal data for 9: C14H14FeO7, M = 350.10, monoclinic, a = 10.1710(4) Å, b = 7.2170(4) Å, c = 10.6320(6) Å, β = 110.426(3)°, V = 731.36(6) Å3, T = 150(2) K, space group P2(1), Z = 2, μ(MoKα) = 1.063 mm−1, 6905 reflections measured, 3713 independent reflections (2θ = 8.57–30.45°, Rint = 0.0685) against 203 parameters gave R1 = 0.0385 and wR2 = 0.1056 [I > 2σ(I)] and R1 = 0.0404 and wR2 = 0.1107 (for all data). The goodness of fit on F2 was 1.054. Flack parameter = −0.003(16).
- W. Yang and M. Koreeda, J. Org. Chem., 1992, 57, 3836 CrossRef CAS; W. A. Szarek, A. Zamojski, K. N. Tiwari and E. R. Ison, Tetrahedron Lett., 1986, 27, 3827 CrossRef CAS.
- A. J. Blacker, R. J. Booth, G. M. Davies and J. K. Sutherland, J. Chem. Soc., Perkin Trans. 1, 1995, 2861 RSC; F. Fabris, J. Collins, B. Sullivan, H. Leisch and T. Hudlický, Org. Biomol. Chem., 2009, 7, 2619 RSC.
- D. R. Boyd, N. D. Sharma, N. I. Bowers, G. B. Coen, J. F. Malone, C. R. O'Dowd, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2010, 8, 1415 RSC and references therein.
- D. A. Owen, A. V. Malkov, I. M. Palotai, C. Roe, E. J. Sandoe and G. R. Stephenson, Chem.–Eur. J., 2007, 13, 4293 CrossRef CAS and references therein.
- A. Watanabe, T. Kamahori, M. Aso and H. Suemune, J. Chem. Soc., Perkin Trans. 1, 2002, 2539 RSC.
- L. Kruczynski and J. Takats, Inorg. Chem., 1976, 15, 3140 CrossRef.
- C. G. Kreiter, S. Stüber and L. Wackerle, J. Organomet. Chem., 1974, 66, C49 CrossRef CAS; L. Kruczynski and J. Takats, J. Am. Chem. Soc., 1974, 96, 932 CrossRef CAS; J.-Y. Lallemand, P. Laszlo, C. Muzette and A. Stockis, J. Organomet. Chem., 1975, 91, 71 CrossRef; D. Liebfritz and H. tom Dieck, J. Organomet. Chem., 1976, 105, 255 CrossRef CAS; K. S. Claire, O. W. Howarth and A. McCamley, J. Chem. Soc., Dalton Trans., 1994, 2615 RSC; H.-J. Knölker, G. Baum, N. Foitzik, H. Goesmann, P. Gonser, P. G. Jones and H. Röttele, Eur. J. Inorg. Chem., 1998, 993 CrossRef.
- For selected examples, see: D. R. Boyd, N. D. Sharma, S. A. Barr, H. Dalton, J. Chima, G. Whited and R. Seemayer, J. Am. Chem. Soc., 1994, 116, 1147 Search PubMed; C. C. R. Allen, D. R. Boyd, H. Dalton, N. D. Sharma, I. Brannigan, N. A. Kerley, G. N. Sheldrake and S. C. Taylor, J. Chem. Soc., Chem. Commun., 1995, 117 CrossRef CAS; T. Hudlický, C. D. Claeboe, L. E. Brammer, Jr., L. Koroniak, G. Butora and I. Ghiviriga, J. Org. Chem., 1999, 64, 4909 RSC; T. Hudlický, U. Rinner, D. Gonzalez, H. Akgün, S. Schilling, P. Siengalewicz, T. A. Martinot and G. R. Pettit, J. Org. Chem., 2002, 67, 8726 CrossRef CAS.
- For selected examples, see: T. Hudlický, H. Luna, J. D. Price and F. Rulin, J. Org. Chem., 1990, 55, 4683 Search PubMed; T. Hudlický, J. D. Price, F. Rulin and T. Tsunoda, J. Am. Chem. Soc., 1990, 112, 9439 CrossRef CAS; M. G. Banwell, P. Darmos, M. D. McLeod and D. C. R. Hockless, Synlett, 1998, 897 CrossRef CAS; M. G. Banwell, D. C. R. Hockless, J. W. Holman, R. W. Longmore, K. J. McRae and H. T. T. Pham, Synlett, 1999, 1491 CAS; M. G. Banwell, C. Chun, A. J. Edwards and M. M. Voegtle, Aust. J. Chem., 2003, 56, 861 CrossRef CAS; H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bisset, D. Ilceski and T. Hudlický, Tetrahedron, 2009, 65, 9862 CrossRef CAS; K. A. B. Austin, J. D. Elsworth, M. G. Banwell and A. C. Willis, Org. Biomol. Chem., 2010, 8, 751 CrossRef CAS.
- C. E. Dietinger, M. G. Banwell, M. J. Garson and A. C. Willis, Tetrahedron, 2010, 66, 5250 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterisation of Mosher's esters. Details of NMR lineshape analysis. Experimental procedures and spectra. Crystallographic data for 9. CCDC 768284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc01169j |
| This journal is © The Royal Society of Chemistry 2011 |