Highly regio- and enantioselective catalytic asymmetric hydroboration of α-substituted styrenyl derivatives†‡
Clément
Mazet
* and
David
Gérard
University of Geneva, Organic Chemistry Department, Quai Ernest Ansermet 30, 1211-Geneva, Switzerland. E-mail: clement.mazet@unige.ch; Fax: +41 (0)22 3793215; Tel: +41 (0)22 3796288
First published on 14th October 2010
Abstract
The catalytic asymmetric hydroboration of a variety of 1,1-disubstituted olefins has been achieved with excellent yields, perfect regioselectivity and in some cases, high levels of enantioselectivity using readily accessible iridium catalyst.
Whether the process is mediated or catalyzed, in most asymmetric transformations involving olefins as prochiral reagents, 1,1-disubstituted olefins systematically stand out as a particularly more challenging substrate subclass.1 This has been attributed to the difficulty of a chiral auxiliary or a chiral catalyst to discriminate between two relatively similar substituents at a geminal position. Consequently, enantioselectivities for these substrates are usually lower than those obtained for the corresponding 1,2-disubstituted, trisubstituted or even tetrasubstituted analogues. Asymmetric hydroboration of terminal olefins is no exception and, for several decades, even the most selective and general chiral hydroborating agents were revealed to be incapable of unraveling this long-standing synthetic problem.2 Recently, Soderquist and coworkers designed chiral variants of 9-borabicyclononane (9-BBN) that mediated the hydroboration of four different terminal olefins displaying unprecedented enantioselectivity levels for the most biased substrates (28–92% ee).3 As a next step in this direction, the development of transition metal-catalyzed hydroboration reactions would provide an attractive alternative to the use of expensive and sacrificial stoichiometric chiral auxiliaries. The strongly favored Markovinov regioselectivity observed with most chiral transition metal catalysts investigated to date has certainly precluded the development of a catalytic asymmetric hydroboration of terminal olefins.4 Suzuki,5 Burgess,6 and Ito and Hayashi7 have independently demonstrated the difficulty of obtaining specific boration at the terminal position (β) rather than at the more substituted position (α) using chiral rhodium catalysts. Moderate β-regioselectivities along with low enantioselectivities were obtained in the best cases. Perhaps not surprisingly, subsequent studies have focused on developing highly α-regioselective chiral rhodium catalysts using monosubstituted styrenyl derivatives as prochiral substrates.8 We present herein the approach we followed towards the discovery of a highly regio- and enantioselective iridium-catalyzed hydroboration of terminal olefins.
Building on an early report by Marder and Baker,9 Crudden10 and Miyaura11 have concomitantly established that iridium catalysts display a complementary regioselectivity to that of rhodium catalysts in the hydroboration of styrenyl derivatives.4,8,12 Interestingly, chiral hydrogenationiridium catalysts rank among the very rare successful examples that impart high levels of enantioselectivity in an asymmetric process employing terminal prochiral alkenes.13 Owing to the mechanistic similarities between hydrogenation and hydroboration, which share the same elementary steps (i.e. oxidative addition, migratory insertion, reductive elimination), we reasoned that iridium would be the transition metal of choice to develop highly regio- and enantioselective catalysts for the asymmetric hydroboration of 1,1-disubstituted olefins.
We began our investigations by evaluating the potential of chiral ligands that have proven successful in the context of iridium-catalyzed asymmetric hydrogenation of various classes of olefins, using α-methylstyrene as test substrate and commercially available [Ir(Cl)(COD)]2 (COD = 1,5-cyclooctadiene). The air-stable pinacolborane (HBpin) was preferred over catecholborane (HBcat) as hydroborating agent, not only because its higher steric demand intrinsically favors β-selectivity but also because it gives access to more robust products.14 Reactions were performed at room temperature, in THF, using 2.5 mol% of in situ generated catalyst (Table 1). Remarkably, only the C1-symmetric phosphinooxazoline ligands (Phox) L7 and L8 provided 2a with encouraging levels of enantioselectivity (32 and 48% ee respectively; Table 1, entries 6 and 7). The use of a cationic iridium precursor led to polymerization of the styrenyl derivative suggesting that the ancillary anionic ligand may play an important role in the hydroboration reaction (Table 1, entry 10).
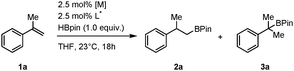
| Entrya | L* | Metal precursor | 2a/3ab | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| a Average of at least two experiments on a 1.0 mmol scale. b Determined by 1H NMR. c Determined by HPLC after conversion to the corresponding alcohol. d Polymerization of 1a was observed. | |||||
| 1 | L1 | [Ir(Cl)(COD)]2 | nd | <5 | nd |
| 2 | L2 | [Ir(Cl)(COD)]2 | nd | <5 | nd |
| 3 | L3 | [Ir(Cl)(COD)]2 | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
75 | <5 |
| 4 | L4 | [Ir(Cl)(COD)]2 | >99:1 |
>99 | <5 |
| 5 | L6 | [[Ir(Cl)(COD)]2 | >99:1 |
99 | <5 |
| 6 | L7 | [Ir(Cl)(COD)]2 | >99:1 |
89 | 32 (S) |
| 7 | L8 | [Ir(Cl)(COD)]2 | >99:1 |
95 | 48 (S) |
| 8 | L9 | [Ir(Cl)(COD)]2 | >99:1 |
55 | 6 (S) |
| 9 | L8 | [Rh(Cl)(COD)]2 | 90:10 |
90 | <5 |
| 10 | L8 | [Ir(COD)2]BArF | nd | nd | ndd |
|
|||||
We next decided to evaluate the influence of a variety of halides and oxyanions on the outcome of the reaction using ligand L8 (Table 2, entries 1–8).15 In all cases, the regioselectivity remained excellent in favor of the β-boration product 2a (2a/3a, >99∶1). Except for X = Br (entry 2), all other anions surveyed led to better enantioselectivities. Although no clear trend is visible with respect to the steric and electronic properties of the anions, notable improvements were obtained in the oxyanions series, with X = OMe offering the best balance in terms of reactivity, regioselectivity and enantioselectivity (Table 2, entry 5). Subsequent solvent optimization studies showed hexanes was the candidate of choice, delivering quantitatively and regioselectively 2a in 92% ee (Table 2, entry 13). Electronic effects were further investigated by subjecting two additional Phox ligands L10 and L11 to the optimized reaction conditions. Introduction of either electron-donating or electron-withdrawing substituents on the para position of the P-aryl rings led to substantially diminished yields and/or enantioselectivities in both cases (Table 2, entries 14 and 15).
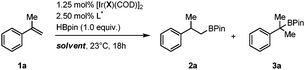
| Entrya | L* | X | Solvent | 2a/3ab | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|---|
| a Average of at least two experiments on a 1.0 mmol scale. b Determined by 1H NMR. c Determined by HPLC after conversion to the corresponding alcohol. | ||||||
| 1 | L8 | Cl | THF | >99:1 |
95 | 48 (S) |
| 2 | L8 | Br | THF | >99:1 |
68 | 30 (S) |
| 3 | L8 | I | THF | >99:1 |
63 | 72 (S) |
| 4 | L8 | OH | THF | >99:1 |
>99 | 60 (S) |
| 5 | L8 | OMe | THF | >99:1 |
>99 | 85 (S) |
| 6 | L8 | Oi-Pr | THF | >99:1 |
>99 | 66 (S) |
| 7 | L8 | Ot-Bu | THF | >99:1 |
75 | 82 (S) |
| 8 | L8 | OPh | THF | >99:1 |
>99 | 72 (S) |
| 9 | L8 | OMe | TBME | >99:1 |
>99 | 88 (S) |
| 10 | L8 | OMe | Acetone | >99:1 |
28 | 71 (S) |
| 11 | L8 | OMe | CH2Cl2 | >99:1 |
66 | 60 (S) |
| 12 | L8 | OMe | Toluene | >99:1 |
97 | 87 (S) |
| 13 | L8 | OMe | Hexanes | >99:1 |
>99 | 92 (S) |
| 14 | L10 | OMe | Hexanes | >99:1 |
99 | 35 (S) |
| 15 | L11 | OMe | Hexanes | >99:1 |
84 | 76 (S) |
To delineate the scope and limitation of our catalytic system, we applied the optimized reaction conditions to a representative set of diversely substituted terminal olefins (Table 3). Introduction of electron-withdrawing substituents on the para or meta positions of model substrate 1a induces a slight but measurable erosion in enantioselectivity (76–80% ee for substrates 1c–1g, Table 3, entries 3–7). A fluorine atom has a stronger negative impact (50% ee, Table 3, entry 2), as much as the introduction of electron-donating substituents for which not only the enantioselectivity, but also the yields are reduced (Table 3, entries 8–10). Increasing the steric demand at the vicinity of the benzylic position by using larger R2alkyl groups (R2 = Et, Cy) or by replacing the phenyl ring with either an o-tolyl or a cyclohexyl drastically reduces the enantioselectivity (Table 3, entries 11–13).
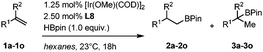
| Entrya | Olefin | R1 | R2 | 2/3b | Yieldc (%) | eed (%) |
|---|---|---|---|---|---|---|
| a Average of at least two experiments on a 1.0 mmol scale. b Determined by 1H NMR. c Isolated yield after chromatography. d Determined by HPLC after conversion to the corresponding alcohol. e Reaction performed at 40 °C. | ||||||
| 1 | 1a | C6H5 | Me | >99:1 |
92 | 92 (S) |
| 2 | 1b | 4-FC6H4 | Me | >99:1 |
83 | 50 (S) |
| 3 | 1c | 4-ClC6H4 | Me | >99:1 |
93 | 77 (S) |
| 4 | 1d | 4-BrC6H4 | Me | >99:1 |
94 | 80 (S) |
| 5 | 1e | 3-BrC6H4 | Me | >99:1 |
91 | 79 (S) |
| 6 | 1f | 4-IC6H4 | Me | >99:1 |
98 | 76 (S) |
| 7 | 1g | 4-(CF3)C6H4 | Me | >99:1 |
92 | 76 (S) |
| 8 | 1h | 4-(MeO)C6H4 | Me | >99:1 |
64 | 32 (S) |
| 9 | 1i | 4-MeC6H4 | Me | >99:1 |
81 | 44 (S) |
| 10 | 1j | 3-MeC6H4 | Me | >99:1 |
88 | 55 (S) |
| 11 | 1k | 2-MeC6H4 | Me | >99:1 |
66 | <5 |
| 12 | 1l | C6H5 | Et | >99:1 |
98 | 31 (S) |
| 13 | 1m | C6H5 | Cy | >99:1 |
55 | 6 (S)e |
Hydroboration of α-methylstyrene using L8 under the optimized reaction conditions and D-Bpin as the hydroborating agent provided interesting preliminary mechanistic insights (Fig. 1). The distribution of deuterium in the product was assessed after oxidation to the corresponding alcohol.16 The high extent of deuterium incorporation in the benzylic position accounts for the expected C(1)-migratory insertion/reductive elimination sequence (A → B). Incorporation of deuterium both in the terminal position and in the methyl group of the substrate is evidence for reversible C(2)-migratory insertion (A → C).
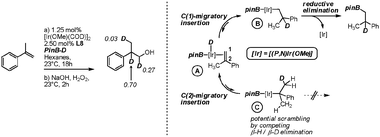 | ||
| Fig. 1 Labelling experiments and mechanistic implications. | ||
The higher extent of incorporation in the terminal position in C can be attributed to the lower entropic cost associated with poising the C(1)-protons rather than the methyl protons in a position favorable for a subsequent β-H-elimination. According to the perfect regioselectivities observed in the hydroboration reactions, reductive elimination from C is virtually not feasible.17 These results also suggest that initial hydride insertion followed by reductive elimination occurs preferentially to boron insertion followed by reductive elimination.
The synthetic applicability of the iridium-catalyzed asymmetric hydroboration of terminal olefins was demonstrated by performing a series of fundamental derivatizations relying on challenging orthogonal functionalization through successive Suzuki cross-coupling reactions (Scheme 1).18
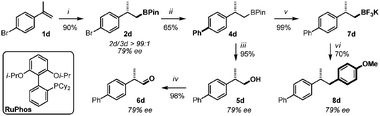 | ||
| Scheme 1 (i) See Table 2 entry 4 (1.0 g scale); (ii) Pd(OAc)2 (10 mol%), dppf (12 mol%), PhB(OH)2 (1.5 equiv.), K3PO4 (3.0 equiv.), THF, 80 °C, 48 h; (iii) NaOH, H2O2, 23 °C, 2 h; (iv) DMP (1.7 equiv.) CH2Cl2, 23 °C, 20 min; (v) KHF2, H2O:MeOH, 23 °C, 30 min; (vi) Pd(OAc)2 (5 mol%), RuPhos (10 mol%), 4-bromoanisole (1.0 equiv.), K2CO3 (3.0 equiv.), toluene:H2O (10:1), 80 °C, 24 h. | ||
Asymmetric hydroboration of 1d was performed on a 1.0 g scale delivering 4d exclusively with excellent yield and good enantioselectivity (90% yield, 79% ee). Using a standard protocol, selective Suzuki cross-coupling between the electrophilic site of 2d and phenylboronic acid was successfully achieved, leaving the boronic ester untouched. No traces of cross-product could be detected and the coupling product 4d was obtained in 65% yield without any extensive optimizations of the reaction conditions. From this pivotal intermediate, successive oxidations, first to the corresponding alcohol 5d and next to the potentially stereo-labile α-chiral aldehyde 6d, were performed without noticeable epimerization of the stereogenic center (93% yield over 2 steps, 79% ee).19 The relatively inert boronate ester 4d was converted to the corresponding potassium trifluoroborate salt 7d in quantitative yield.20 A second Suzuki cross-coupling was conducted exploiting conditions developed by Molander21 and coworkers. Using 5 mol% of Pd(OAc)2 and 10 mol% of Buchwald's RuPhos ligand,224-bromoanisole was coupled with 7d under aqueous conditions. The expected chiral polyaromatic product 8d was obtained in 70% yield without erosion of the enantiomeric purity, demonstrating that competing reversible β-hydride elimination does not occur under these conditions.23
In conclusion, we have developed an efficient protocol for the catalytic asymmetric hydroboration of terminal olefins. High catalytic activity, perfect regioselectivity and enantioselectivities up to 92% were obtained using (phosphinooxazoline)–iridium catalysts. The applicability of this method was further highlighted by orthogonal functionalization through successive Suzuki cross-coupling reactions.
This work was supported by the State Secretariat for Education and Research. Solvias is acknowledged for the gift of ligand L2 and Johnson Matthey for the loan of iridium precursors. Professor E.-P. Kündig is warmly thanked for giving us access to his analytical facilities. L. Mantilli, S. Torche and D. Grassi are acknowledged for practical assistance.
Notes and references
- S. P. Thomas and V. K. Aggarwal, Angew. Chem., 2009, 121, 1928 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1896 ) CrossRef.
- (a) A. Pelter, K. Smith and H. C. Brown, Borane Reagents, Academic Press, New York, 1988 Search PubMed; (b) Y. Bubnov, in Science of Synthesis, ed. D. E. Kaufmann and D. S. Matteson, Thieme, Stuttgart, 2004, vol. 6, pp. 945–1072 Search PubMed.
- A. Z. Gonzalez, J. G. Romàn, E. Gonzalez, J. Martinez, J. R. Medina, K. Matos and J. A. Soderquist, J. Am. Chem. Soc., 2008, 130, 9218 CrossRef CAS.
- (a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179 CrossRef CAS; (b) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695 CrossRef CAS; (c) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957 CrossRef CAS; (d) C. M. Crudden, B. W. Glasspoole and C. J. Lata, Chem. Commun., 2009, 6704 RSC.
- M. Sato, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1990, 31, 231 CrossRef CAS.
- (a) K. Burgess and M. J. Ohlmeyer, J. Org. Chem., 1988, 53, 5178 CrossRef CAS; (b) K. Burgess, W. A. van der Donk and M. J. Ohlmeyer, Tetrahedron: Asymmetry, 1991, 2, 613 CrossRef CAS.
- T. Hayashi, Y. Matsumoto and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 601 CrossRef CAS.
- (a) T. Hayashi, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 1, pp. 351–366 Search PubMed; (b) J. M. Brown, in Modern Rhodium-Catalyzed Organic reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- S. A. Westcott, T. B. Marder and R. T. Baker, Organometallics, 1993, 12, 975 CrossRef CAS.
- C. M. Crudden, Y. B. Hleba and A. C. Chen, J. Am. Chem. Soc., 2004, 126, 9200 CrossRef CAS.
- Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695 CrossRef CAS.
- (a) D. Noh, H. Chea, J. Ju and J. Yun, Angew. Chem., 2009, 121, 6178 ( Angew. Chem., Int. Ed. , 2009 , 48 , 6062 ) CrossRef; (b) C. J. Lata and C. M. Crudden, J. Am. Chem. Soc., 2010, 132, 131 CrossRef CAS.
- (a) S. McIntyre, E. Hörmann, F. Menges, S. P. Smidt and A. Pfaltz, Adv. Synth. Catal., 2005, 347, 282 CrossRef CAS; (b) M. Dièguez, J. Mazuela, O. Pàmies, J. J. Verendel and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 7208 CrossRef CAS; (c) J. Mazuela, J. Verendel, M. Coll, B. Schäffner, A. Börner, P. G. Andersson, O. Pàmies and M. Dièguez, J. Am. Chem. Soc., 2009, 131, 12344 CrossRef CAS.
- A. C. Chen, L. Ren and C. M. Crudden, J. Org. Chem., 1999, 64, 9704 CrossRef CAS.
- K. Fagnou and M. Lautens, Angew. Chem., 2002, 114, 26 ( Angew. Chem., Int. Ed. , 2002 , 41 , 26 ) CrossRef.
- D. A. Evans, G. C. Fu and B. A. Anderson, J. Am. Chem. Soc., 1992, 114, 6679 CrossRef CAS.
- The formation of π-allyl iridium intermediates can be ruled out because it would imply the formation of coordinatively over-saturated iridium complexes. An achiral ligand gives the same isotopic distribution.
- (a) M. Tobisu and N. Chatani, Angew. Chem., 2009, 121, 3617 ( Angew. Chem., Int. Ed. , 2009 , 48 , 3565 ) CrossRef; (b) N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS.
- C. Botuha, M. Haddad and M. Larchevêque, Tetrahedron: Asymmetry, 1998, 9, 1929 CrossRef CAS.
- (a) S. Darses and J.-P. Genet, Eur. J. Org. Chem., 2003, 4313 CrossRef CAS; (b) V. Bagutski, A. Ros and V. K. Aggarwal, Tetrahedron, 2009, 65, 9956 CrossRef CAS.
- G. A. Molander and L. Jean-Gérard, J. Org. Chem., 2009, 74, 5446 CrossRef CAS.
- R. Martín and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS.
- Y. Iwai, K. M. Gligorich and M. S. Sigman, Angew. Chem., 2008, 120, 3263 ( Angew. Chem., Int. Ed. , 2008 , 47 , 3219 ) CrossRef.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Full experimental data and characterization for all new compounds. See DOI: 10.1039/c0cc01547d |
| This journal is © The Royal Society of Chemistry 2011 |