Modifiable bidentate systems via N–C rearrangement in triazoles†‡
Elana A. Slutsky
Smith
,
Gregory
Molev
,
Mark
Botoshansky
and
Mark
Gandelman
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: chmark@tx.technion.ac.il; Fax: +972 4-829-5703
First published on 23rd August 2010
Abstract
A new N–C rearrangement of substituted triazoles has been discovered. This process has been applied to the synthesis of diverse classes of mixed bidentate ligands and their metal complexes with highly modifiable backbones, including the first bisphosphine zwitterionic system based on a triazole frame.
Recently, we reported on the first combinatorial approach towards the synthesis of tridentate ligands.1 This methodology was based upon the Cu(I) catalyzed [2+3] cycloaddition of alkynes and azides, forming 1,4-substituted triazoles, as the general strategy for ligand assembly.2–4 Selective combinatorial preparation of mixed bidentate ligands by covalent assembly of two monomeric donor units would additionally be of high interest.5 Remarkably, while 1,4-substituted triazoles prepared by click chemistry are widely used for molecular assembly, the complementary regioisomer, the 1,5-substituted triazole, is practically unemployed for building functional compounds with completely different molecular geometry. We hypothesized that selective synthesis of 1,5-substituted triazolesvia reaction of metal acetylides with organic azides could be a useful building tool for this purpose (Scheme 1).6 Here we report that, in the course of our studies, we have discovered a new N–Crearrangement of substituted triazoles, mediated by phosphine oxide. Although N–Nrearrangements in triazoles are documented, the N–C counterparts are yet unprecedented.7 Based on this rearrangement, bisphosphine ligands, possessing a neutral or negatively charged highly modifiable triazole backbone, and their metal complexes were prepared for the first time.8 Among these complexes is an unprecedented triazole-derived bisphosphine zwitterionic metal system.
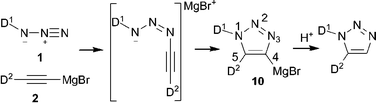 | ||
| Scheme 1 Mechanism for selective synthesis of 1,5-substituted triazoles. | ||
The envisioned synthetic route for ligand preparation (including a previously proposed mechanism) is presented in Scheme 1.6 Our approach was to combine azides 1 and acetylides 2, each decorated with a monomeric ligand unit (D1 and D2), in order to directly form desired mixed bidentate systems in a combinatorial manner. In order to check our hypothesis, the prepared alkyne 3 was converted to acetylide 4 and then reacted in situ with azide 5a (Scheme 2).9 To our surprise, the single isolated product of this reaction was the 4,5-substituted triazole 6a rather than the expected 1,5-substituted isomer 7a. The 31P NMR spectrum of 6a exhibits a singlet at 18 ppm for (Ph)2P(O) and a multiplet (due to P–B coupling) at 31 ppm for (i-Pr)2P(BH3). Both signals are in the range characteristic of a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (E = BH3 or O) groupbound to carbon. The P–N pattern expected for the 1,5-isomer 7a would typically resonate at ∼70 ppm in 31P NMR.10 The two carbons of the triazole appear as doublets in 13C NMR due to the coupling with P-based substituents. The molecular structure of 6a was confirmed by X-ray analysis of its single crystals (Fig. 1).11 Thus, the net result of this reaction is the transfer of the phosphine group from the 1-N to the 4-C position of the triazole ring.
E (E = BH3 or O) groupbound to carbon. The P–N pattern expected for the 1,5-isomer 7a would typically resonate at ∼70 ppm in 31P NMR.10 The two carbons of the triazole appear as doublets in 13C NMR due to the coupling with P-based substituents. The molecular structure of 6a was confirmed by X-ray analysis of its single crystals (Fig. 1).11 Thus, the net result of this reaction is the transfer of the phosphine group from the 1-N to the 4-C position of the triazole ring.
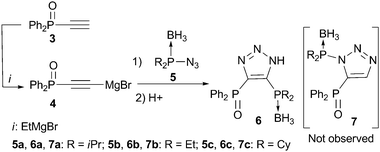 | ||
| Scheme 2 Synthesis of bisphosphines 6viarearrangement. | ||
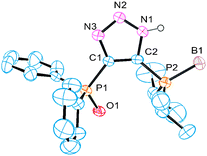 | ||
| Fig. 1 Perspective view of molecule 6a. Hydrogen atoms (except N–H) are omitted for clarity. | ||
Remarkably, when phenyl acetylene (8a) or diphenylethynylphosphine borane complex 8b (which differs from 3 only in the phosphine protecting group) was reacted with azide 5a under conditions identical to the previous experiment, non-rearranged 1,5-substitued triazoles 9 were obtained (Scheme 3). Signals of these products in 31P NMR at 71 and 73 ppm, for 9a and 9b correspondingly, are typical for phosphorus substituted with a nitrogen atom ((iPr)2P(BH3)–N fragment; for comparison of 31P NMR spectra of rearranged and non-rearranged products 6a and 9 see figure in ESI).‡10 The lack of N-to-C group transfer in these control experiments indicates that the phosphine oxide group of 3 is crucially involved in the rearrangement process illustrated in Scheme 2. The observed formation of 9 points toward an intermediate of type 10 (formed in the course of the reaction of 3 with 5; Scheme 1) as the most probable species to undergo rearrangement.12
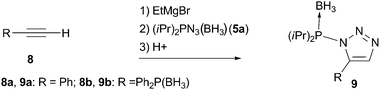 | ||
| Scheme 3 No rearrangement observed in reactions with acetylides lacking phosphine-oxide moiety. | ||
The driving force of this rearrangement is apparently the formation of the stronger P–C and N–Mg bonds rather than the respectively weaker P–N and C–Mg bonds. Indeed, DFT calculations at b31yp/6-31+g(d,p) level of theory13 show that such bond reorganization is favorable by 45.6 kcal mol−1 for 11a (Scheme 4). Interestingly, such rearrangement for species 11b is equally enthalpically favored. The same thermodynamic driving force for 11a and 11b (the analogue of which does not, in practice, undergo N-to-C transfer) supports the involvement of the phosphine oxide unit in this rearrangement. Detailed mechanistic studies of this rearrangement as well as further applications of phosphine oxides in related activation/transfer processes are under thorough investigation in our labs and will be published elsewhere.
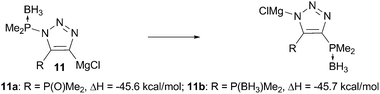 | ||
| Scheme 4 DFT calculations for P–C vs. P–N bond formation. | ||
This new rearrangement is not limited to azide 5a. For example, phosphinoazides 5b and 5c also undergo this process to furnish compounds 6b and 6c, respectively (Scheme 2). The fastest and most facile reaction was observed for 5b, which bears the least sterically congested ethyl substituents on the phosphorus atom (the isolated yield was 85%).
Compounds 6 represent very attractive substrates for the synthesis of diverse families of bidentate ligands for transition metals. When refluxed in methanol, 6a undergoes selective deboranation to result in the novel triazole-based bisphosphine monooxide ligand 12 (Scheme 5). This compound on its own is of particular interest, since bidentate ligands bearing hard–soft donor pairs have found spectacular applications in catalysis.14 Compound 12 is reduced to give the non-symmetrical bisphosphine ligand 13 in excellent yield. The triazole frame can be further functionalized by selective deprotonation of the N–H to furnish the anionic non-symmetrical ligand 14, or by N-substitution to form the neutral alkylated ligand 16 (Scheme 5).
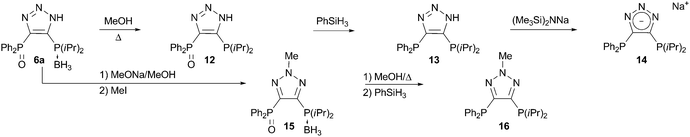 | ||
| Scheme 5 Synthesis of ligands 12, 13, 14, and 16. | ||
Due to our interest in mixed soft ligands, we investigated the coordination behavior of bisphosphines 13, 14, and 16 with late transition metals. Ligand 13 did not demonstrate selective chelation and irresolvable mixtures of products were usually obtained upon its reaction with various metal precursors. This is likely due to the relatively acidic N–H bond of the triazole. Indeed, to our delight, both anionic 14 and methylated neutral counterpart 16 exhibited selective behavior with late transition metals and their coordination mode depends on the charge of the backbone. When ligand 16 reacts with (COD)PtCl2 (COD = cyclooctadiene), exclusive formation of complex 17 is observed (Scheme 6). The 31P NMR spectrum of 17 shows two doublets at 1.8 and 35.1 ppm with a typical cis phosphorus–phosphorus coupling constant of 6.9 Hz. 195Pt–P satellites for both doublets with JPPt = 3741 and 3542 Hz, respectively, clearly indicate coordination of both phosphine arms to the platinum center. An X-ray analysis of single crystals of 17 shows a distorted square planar structure with P–Pt–P bite angle of 89.81(0.12)° (Fig. 2a).
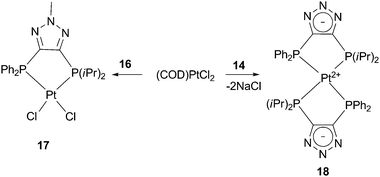 | ||
| Scheme 6 Type of complex formed is dependent upon ligand charge. | ||
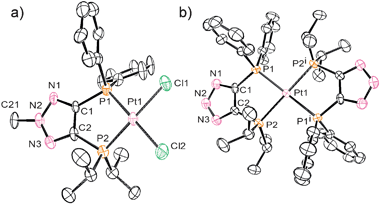 | ||
| Fig. 2 Perspective view of molecules (a) 17 and (b) 18. | ||
Completely different reactivity was observed for the negatively charged ligand 14. Quantitative formation of zwitterionic complex 18 was obtained upon reaction of 14 with the same platinum precursor (Scheme 6). The driving force for the formation of this bis-diphosphine compound is apparently the precipitation of NaCl. The 31P NMR spectrum of 18 exhibits two triplet sets with 195Pt–P satellites which indicate binding of two ligands per platinum center. The structure of 18 was confirmed by X-ray analysis (Fig. 2b). The platinum atom is located at the intersection of the two five-membered chelate rings and the whole molecule has C2 symmetry. Compound 18 represents the first example of a zwitterionic bisphosphine complex based on a triazole frame. Although examples of mesoionic or “truly zwitterionic” metal complexes bearing chelating bidentate ligands with negatively charged backbones are limited, they have found spectacular applications in inorganic and organometallic chemistry.15,16 As such, complexes based on anionic ligand 14 represent an attractive extension of this family of compounds.
The prepared ligand set allows for direct access to both neutral and zwitterionic metal complexes. Since ligands 14 and 16 are isostructural and isoelectronic, they open the door to direct evaluation of the influence of ligand-charge on the properties of the metal center. Moreover, our zwitterionic system offers an interesting alternative to the known bisphosphine boronate-based zwitterionic complexes, as the charged triazolide backbone (possessing an N− unit) can be further functionalized or can serve as an additional reaction center.16
In summary, we have discovered a new N–C rearrangement of substituted triazoles which represents a valuable extension of the chemistry of these increasingly useful compounds. This allowed us to prepare novel diverse classes of bisphosphine ligands and their metal complexes with highly modifiable triazole backbones. These include the first zwitterionic complex prepared based on a bisphosphine triazole frame. Investigation of the properties of these systems, especially of the potential for cooperative chemistry between the basic/nucleophilic negatively charged triazolide ligand and the metal center, is currently underway in our labs.
Financial support from Israel Science Foundation (Grant No. 1292/07) and The FIRST Program of the Israel Science Foundation (Grant No. 1514/07) is acknowledged.
Notes and references
- E. M. Schuster, M. Botoshansky and M. Gandelman, Angew. Chem., Int. Ed., 2008, 47, 4555–4558 CrossRef CAS; E. M. Schuster, G. Nisnevich, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 5025 CrossRef CAS.
- A. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS; P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7 CAS.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- For review on use of the Cu(I)-catalyzed azide–alkyne cycloaddition for adaptable ligands construction, see: H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 Search PubMed.
- For representative examples of non-covalent combinatorial approaches for synthesis of bidentate ligands, see: B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816 Search PubMed; C. Waloch, J. Wieland, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2007, 46, 3037 CrossRef CAS; A. C. Laungani and B. Breit, Chem. Commun., 2008, 844 CrossRef CAS; V. F. Slagt, M. Röder, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 4056 RSC; P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162 CrossRef CAS; J. M. Takacs, D. S. Reddy, S. A. Moteki, D. Wu and H. Palencia, J. Am. Chem. Soc., 2004, 126, 4494 CrossRef CAS . For solid phase-based approach, see: G. Y. Li, P. J. Fagan and P. L. Watson, Angew. Chem., Int. Ed., 2001, 40, 1106 CrossRef CAS.
- G. S. Akimova, V. N. Chistokletov and A. A. Petrov, Zh. Org. Khim., 1967, 3, 968 CAS; A. Krasinski, V. V. Fokin and K. B. Sharpless, Org. Lett., 2004, 6, 1237 CrossRef.
- S. W. Kwok, J. E. Hein, V. V. Fokin and K. B. Sharpless, Heterocycles, 2008, 76, 1141 CrossRef CAS; M. Yamauchi, T. Miura and M. Murakami, Heterocycles, 2010, 80, 177 CrossRef CAS.
- Sole example of symmetrical 4,5-bis(diphenylphosphinoyl)-1,2,3-triazole and its seleno-analog were previously prepared by thermal Huisgen cycloaddition of the corresponding Ph2P(E)C≡CP(E)Ph2 (E = O, Se) with NaN3 and studied as ligands for transition metals. However, metal complexes based on the reduced bis(diphenylphosphanyl)-1,2,3-triazole are not reported. See: A. L. Rheingold, L. M. Liable-Sands and S. Trofimenko, Angew. Chem., Int. Ed., 2000, 39, 3321–3324 Search PubMed; M. Moya-Cabrera, V. Jancik, R. A. Castro, R. Herbst-Irmer and H. W. Roesky, Inorg. Chem., 2006, 45, 5167 CrossRef CAS; J. A. Balanta-Díaz, M. Moya-Cabrera, V. Jancik, L. W. Pineda-Cedeño, R. A. Toscano and R. Cea-Olivares, Inorg. Chem., 2009, 48, 2518 CrossRef CAS; J. Alcántara-Garcia, V. Jancik, J. Barroso, S. Hidalgo-Bonilla, R. Cea-Olivares, R. A. Toscano and M. Moya-Cabrera, Inorg. Chem., 2009, 48, 5874 CrossRef CAS; S. Trofimenko, A. L. Rheingold and C. D. Incarvito, Angew. Chem., Int. Ed., 2003, 42, 3506 CrossRef CAS.
- See ESI‡ for experimental details.
- For some relevant examples of 31P NMR spectra of (Alk)2P(E)–N (E = BH3 or O) pattern, see: C. G. Hartung, A. Fecher, B. Chapell and V. Snieckus, Org. Lett., 2003, 5, 1899 Search PubMed; R. K. Kanjolia, D. K. Srivastava, C. L. Watkins and L. K. Krannich, Inorg. Chem., 1989, 28, 3341 CrossRef CAS; B. Wrackmeyer, Spectrochim. Acta, Part A, 1984, 40, 963 CrossRef CAS.
- CCDC 767176 (6a), 767178 (17) and 767177 (18) contain the supplementary crystallographic data for this paper (ESI‡).
- Once intermediate 10 is formed, PO in this species may act as a Lewis base activator of the electrophilic iPr2P(BH3) group for nucleophilic attack by a second molecule of 10. Activation of Lewis acids with Lewis basic phosphine oxides is well documented; see: S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 Search PubMed , and references therein; X. Pu, X. Qi and J. M. Ready, J. Am. Chem. Soc., 2009, 131, 10364 CrossRef CAS . Another possibility is intramolecular transfer of the iPr2P(BH3) group by double nucleophilic displacement, initiated by PO, and involving a (Ph)2P+–O–P(iPr)2 species as an intermediate.
- Calculated using Gaussian 03 (Revision D.01): M. J. Frisch, et al., see ESI‡.
- A. B. Charette, A. A. Boezio, A. Côté, E. Moreau, J. Pytkowicz, J.-N. Desrosiers and C. Legault, Pure Appl. Chem., 2005, 77, 1259–1267 CrossRef CAS; N. J. Farrer, R. McDonald, T. Piga and J. S. McIndoe, Polyhedron, 2010, 29, 254 CrossRef CAS.
- R. Chauvin, Eur. J. Inorg. Chem., 2000, 577 CrossRef CAS; M. Stradiotto, K. D. Hesp and R. J. Lundgren, Angew. Chem., Int. Ed., 2010, 49, 494 CrossRef CAS.
- For some related examples, see: J. C. Thomas and J. C. Peters, J. Am. Chem. Soc., 2001, 123, 5100 Search PubMed; C. C. Lu and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 15818 CrossRef CAS; A. N. Vedernikov, J. C. Fettinger and F. Mohr, J. Am. Chem. Soc., 2004, 126, 11160 CrossRef CAS; E. Khaskin, P. Y. Zavalij and A. N. Vedernikov, Angew. Chem., Int. Ed., 2007, 46, 6309 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, X-ray crystallographic data and DFT calculations. CCDC 767176 (6a), 767178 (17) and 767177 (18). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02033h |
| This journal is © The Royal Society of Chemistry 2011 |