A new tetrathiafulvalene–phenoxynaphthacenequinone dyad: switching on the intramolecular electron-transfer with UV light irradiation and metal ion coordination†‡
Lina
Jia
,
Guanxin
Zhang
*,
Deqing
Zhang
*,
Junfeng
Xiang
,
Wei
Xu
and
Daoben
Zhu
Beijing National Laboratory for Molecular Sciences, Organic Solids Laboratory, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: dqzhang@iccas.ac.cn
First published on 19th August 2010
Abstract
The metal ion-promoted intramolecular electron transfer within TTF-based dyad 1 containing a photochromic PNQ unit can be switched on with UV light irradiation by taking advantage of the photochromic feature of the PNQ unit and the fact that the trans and ana forms of PNQ show different electron accepting capacity.
As strong electron donors TTF (tetrathiafulvalene) and its derivatives have been widely employed as very important building blocks for organic conducting materials, molecular machines, fluorescence switches and chemical sensors.1–4 Among them, TTF-based D–A systems have been extensively studied for the intramolecular charge transfer/photoinduced electron transfer processes.2,3 We have recently reported the metal ion-promoted electron transfer within TTF–quinone dyads in which the TTF and quinone units are covalently linked by an oligoethylene glycol chain.4 The investigations manifest that the coordination of the oligoethylene glycol chain and the radical anion of quinone with metal ions is crucial for facilitating the electron transfer. Further studies indicate that such metal ion-promoted electron transfer is dependent on the electron accepting capacities of the quinone units of the TTF–quinone dyads;4b the stronger an acceptor the quinone unit is, the more efficient the intramolecular electron transfer becomes for the TTF–quinone dyad in the presence of certain metal ions. Accordingly, it would be possible to tune the intramolecular electron transfer within TTF–quinone dyads by varying the electron accepting capacities of the quinone units.
Phenoxynaphthacenequinone (PNQ), as a typical photochromic compound, undergoes photoisomerization from ‘trans’ form to ‘ana’ form upon UV light irradiation and the reverse conversion occurs upon further visible light irradiation as illustrated in Scheme 1.5 More importantly, the ‘ana’ form of PNQ is a stronger electron acceptor compared to the corresponding ‘trans’ form.5c,e This is also confirmed by theoretical calculations (see ESI); the LUMO energy of the ‘ana’ form (−3.02 eV) is lower than that of the corresponding ‘trans’ form (−2.58 eV). Thus, the electron accepting capacity of PNQ can be tuned by UV/visible light irradiation. Accordingly, it is interesting to examine the TTF–PNQ dyad and it is anticipated that the intramolecular electron transfer within the TTF–PNQ dyad in the presence of metal ions can be tuned by light irradiation. With these considerations in mind the TTF–PNQ dyad 16 (Scheme 1) in which the TTF and PNQ units are linked by a glycol chain was designed and investigated. The absorption and ESR spectral investigations clearly manifest that intramolecular electron transfer takes place in dyad 1 after cascade input of UV light (310 nm) and metal ions (Sc3+, Pb2+ and Zn2+).
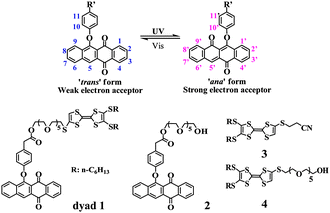 | ||
| Scheme 1 Photoisomerization of phenoxynaphthacenequinone and chemical structures of compounds 1–4. | ||
The syntheses of dyad 1 and relevant compounds are deposited in ESI. Fig. 1 shows the absorption spectrum of dyad 1 and those after UV light irradiation for different times. Dyad 1 exhibits weak absorptions above 400 nm. After UV light irradiation new absorption bands around 450 nm and 479 nm emerged gradually as depicted in Fig. 1. These new absorptions are due to the ‘ana’ form of the PNQ unit in dyad 1, which was generated from the ‘trans’ form of the PNQ unit upon UV light irradiation according to previous reports.5 A control experiment with compound 2 under UV light irradiation also confirms this assumption. The photostationary state was achieved after UV light irradiation for 110 s. Further visible light (500 nm) induced a decrease for the absorption bands around 450 nm and 479 nm and the absorption spectrum of dyad 1 can be restored after further visible light irradiation for 100 s as shown in Fig. S1. The inset of Fig. 1 demonstrates the reversible absorbance variation at 479 nm for dyad 1 after alternating UV and visible light irradiation for several cycles. Thus, the ‘trans’ form and the ‘ana’ form of the PNQ unit in dyad 1 can be reversibly transformed by UV and visible light irradiation.7
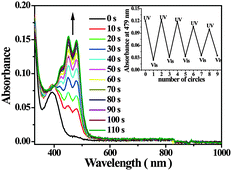 | ||
| Fig. 1 Absorption spectra of dyad 1 in degassed CH2Cl2 (1.0 × 10−5 M) after UV light (310 nm) irradiation for different times; inset shows the variation of absorbance at 479 nm for the solution of dyad 1 after alternating UV and visible light irradiation. | ||
The photoisomerization of the PNQ unit in dyad 1 was investigated with 1H-NMR spectroscopy. As shown in Fig. S3, the signals due to the aromatic protons of dyad 1 were changed clearly after UV light irradiation. For example, the signals of H10 (see Scheme 1, 6.83 ppm) and H11 (7.20 ppm) became weak gradually and two new signals at 7.00 ppm and 7.23 ppm emerged upon UV light irradiation. The molar ratio of the ‘ana’ form and the ‘trans’ form of the PNQ unit of dyad 1 in the photostationary state was ca. 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 based on the 1H-NMR data. Similarly, the molar ratio of the ‘ana’ form and the ‘trans’ form of compound 2 in the photostationary state was estimated to be ca. 6:1 under the same conditions.
:3 based on the 1H-NMR data. Similarly, the molar ratio of the ‘ana’ form and the ‘trans’ form of compound 2 in the photostationary state was estimated to be ca. 6:1 under the same conditions.
Further examination of the absorption spectrum of dyad 1 and that after UV light irradiation indicates that the interaction between the TTF and PNQ (both ‘trans’ form and ‘ana’ form) units within the dyad can be neglected. The absorption spectrum of dyad 1 is almost the sum of those of compounds 2 and 3 (see Fig. S4). Similarly, the absorption spectrum of dyad 1 after UV light irradiation contains mainly the absorptions of the TTF unit and the ‘ana’ form of the PNQ unit (see Fig. 1). Moreover, the cyclic voltammetric investigations also manifest that the interaction between the TTF and PNQ (both ‘trans’ form and ‘ana’ form) units within the dyad can be negligible. Dyad 1 exhibits two quasi-reversible oxidation waves with E1/2ox1 = 0.55 V and E1/2ox2 = 0.93 V, as shown in Fig. S5, which are rather close to those of compounds 3 and 4.8 After UV light irradiation the two oxidation potentials due to the TTF unit (E1/2ox1 = 0.55 V and E1/2ox2 = 0.93 V) keep almost unaltered. However, a new reduction wave at −0.74 V appears (see Fig. S5), which should be due to the ‘ana’ form of the PNQ unit in dyad 1 according to previous reports.5
In the following we demonstrate that intramolecular electron transfer occurs within dyad 1 in the presence of metal ions after UV light irradiation. Addition of metal ions such as Pb2+ and Sc3+ to the solution of dyad 1 led to almost no absorption spectral change (see Fig. S6). However, when the solution of dyad 1 was exposed to UV light for a certain time, new absorptions due to the TTF˙+ were detected for dyad 1 after the addition of metal ions. For instance, new absorption around 790 nm emerged gradually after the addition of Pb2+ to the solution of dyad 1 which was irradiated with UV light (310 nm) for 110 s as depicted in Fig. 2A; moreover, the intensity of this new absorption was increased by increasing the amounts of Pb2+ added to the solution and maximum absorption was reached after the addition of 5.0 eq. of Pb2+. Additionally, the absorption around 450 nm was also slightly enhanced after introducing Pb2+ to the solution of dyad 1. According to previous studies,4,9 the absorption bands around 450 and 790 nm can be ascribed to the formation of the radical cation of the TTF unit (TTF˙+) in dyad 1. In fact, this conclusion was supported by the observation of new absorptions around 450 nm and 790 nm for reference compound 3 after the direct oxidation with 1.0 eq. of Fe3+ (see Fig. S7). It should be noted that a similar absorption spectral change was detected for dyad 1 after addition of Pb2+ and further UV light irradiation (Fig. S10). These absorption spectral investigations clearly indicate that the TTF unit within dyad 1 in which the PNQ unit exists in the ‘ana’ form was oxidized, i.e. intramolecular electron transfer can take place within dyad 1 after the addition of Pb2+.
![(A) Absorption spectra of dyad 1 recorded in degassed CH2Cl2 (1.0 × 10−5 M) after UV (310 nm) irradiation for 110 s and further addition of different amounts of Pb2+ [Pb(ClO4)2]. (B)The ESR spectrum of dyad 1 (1.0 × 10−3 M) in CH2Cl2 after UV light (310 nm) irradiation for 25 min in the presence of 5.0 eq. of Pb2+ [Pb(ClO4)2] recorded at room temperature; the solution was degassed before measurement.](/image/article/2011/CC/c0cc02110e/c0cc02110e-f2.gif) | ||
| Fig. 2 (A) Absorption spectra of dyad 1 recorded in degassed CH2Cl2 (1.0 × 10−5 M) after UV (310 nm) irradiation for 110 s and further addition of different amounts of Pb2+ [Pb(ClO4)2]. (B)The ESR spectrum of dyad 1 (1.0 × 10−3 M) in CH2Cl2 after UV light (310 nm) irradiation for 25 min in the presence of 5.0 eq. of Pb2+ [Pb(ClO4)2] recorded at room temperature; the solution was degassed before measurement. | ||
Furthermore, strong ESR signals were detected for dyad 1 after UV light irradiation and addition of Pb2+ as depicted in Fig. 2B. The solution of dyad 1 in the presence of 5.0 eq. of Pb2+ after UV light irradiation exhibited strong ESR signals (doublet, g = 2.01001). The ESR signals are likely due to the radical cation of the TTF unit in dyad 1 according to previous studies.4 In comparison, no ESR signals were observed for dyad 1 in the presence of Pb2+ without UV light irradiation. These ESR data also indicate that intramolecular electron transfer occurs within dyad 1 after sequential UV light irradiation and the addition of Pb2+.
Besides Pb2+, other metal ions were also examined with regard to the intramolecular electron transfer within dyad 1. Both absorption and ESR studies (see Fig. S8–S9) show that Sc3+ and Zn2+ can also induce the intramolecular electron transfer between the TTF and PNQ (as ana form) units of dyad 1. New absorptions around 450 nm and 790 nm were observed for dyad 1 in the presence of either Zn2+ or Sc3+ after UV light irradiation (see Fig. S8). Similarly, ESR signals were detected for dyad 1 after UV light irradiation and addition of either Zn2+ or Sc3+ (see Fig. S9). The above results clearly indicate that metal ion-promoted electron transfer cannot take place within dyad 1 before UV light irradiation, but it occurs efficiently after UV light irradiation as schematically illustrated in Scheme 2. Therefore, the intramolecular electron transfer within dyad 1 in the presence of metal ions can be tuned with UV light irradiation.
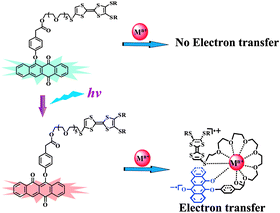 | ||
| Scheme 2 Illustration of the mechanism for the intramolecular electron transfer after UV light irradiation and introducing metal ions. | ||
In order to understand the metal ion-promoted electron transfer within dyad 1 after UV light irradiation, the cyclic voltammograms of reference compound 2 in the presence of metal ions (Pb2+ and Sc3+) after UV light irradiation were recorded as depicted in Fig. S11. The ‘ana’ form of 2 generated after UV light irradiation showed a reduction peak at −0.61 V. Interestingly, the reduction peak was positively shifted after introducing either Pb2+ or Sc3+. For instance, the reduction peak potential of the ‘ana’ form of 2 (generated after UV light irradiation for 25 min) was shifted from −0.61 V to −0.52 V in the presence of 5.0 eq. of Sc3+. In the presence of Sc3+, the cyclic voltammograms of 2 after UV light irradiation for 25 min were recorded at different scanning rates. By following Fukuzumi’s method,10 the reduction potential of the ana form of 2 was estimated to be −0.47 V in the presence of 5.0 eq. of Sc3+ (Fig. S11–S12). Therefore, it can be concluded that the electron accepting capacity of the ‘ana’ form of PNQ can be enhanced in the presence of metal ions. Accordingly, it is anticipated that the electron transfer between the TTF and PNQ (‘ana’ form) units in dyad 1 would become more feasible in the presence of metal ions. But, the electron transfer is still not thermodynamically favorable by considering the oxidation potential of the TTF unit (E1/2ox1 = 0.55 V). As for the metal ion-promoted electron transfer within TTF–quinone dyads, we propose that oxygen atoms of the oligoethylene glycol chain, oxygen atoms of the radical anion of the PNQ (‘ana’ form) unit and the sulfur atom from TTF˙+ in dyad 1 may coordinate with Pb2+/Sc3+/Zn2+ to stabilize the electron-transfer state by increasing the interaction between the corresponding cation and anion (see Scheme 2).11 It should be mentioned that the electron accepting capacity of PNQ (trans form) before UV light irradiation is rather weak,5c,e and an obvious reduction peak cannot be detected even in the presence of metal ions. Therefore, it is understandable that intramolecular electron transfer within dyad 1 cannot take place in the presence of metal ions before UV light irradiation.
In summary, a new TTF-based dyad 1 with a photochromic PNQ unit was synthesized and studied. Both absorption and ESR spectral investigations manifest that intramolecular electron transfer within dyad 1 in the presence of Pb2+/Sc3+/Zn2+ cannot occur, but it can take place after UV light irradiation. Therefore, the metal ion-promoted electron transfer within dyad 1 is switched on with UV light irradiation by taking advantage of the photochromic feature of the PNQ unit and the fact that the ‘trans’ and ‘ana’ forms of PNQ show different electron accepting capacity.
Notes and references
- (a) N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527 CrossRef CAS; (b) M. R. Bryce, Adv. Mater., 1999, 11, 11 CrossRef CAS; (c) J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS; (d) J. O. Jeppesen, M. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115 CrossRef CAS; (e) N. Martín, L. Sánchez, M. A. Herranz, B. Illescas and D. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS; (f) W. Dichtel, O. Miljanić, W. Zhang, J. Spruell, K. Patel, I. Aprahamian, J. Health and J. Stoddart, Acc. Chem. Res., 2008, 41, 1750 CrossRef CAS; (g) J. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC; (h) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC , and further references therein.
- (a) D. F. Perepichka, M. R. Bryce, C. Pearson, M. C. Petty, E. J. L. McInnes and J. P. Zhao, Angew. Chem., Int. Ed., 2003, 42, 4636 CrossRef CAS; (b) E. Tsiperman, J. Y. Becker, V. Khodorkovsky, A. Shames and L. Shapiro, Angew. Chem., Int. Ed., 2005, 44, 4015 CrossRef CAS; (c) J. Baffreau, F. Dumur and P. Hudhomme, Org. Lett., 2006, 8, 1307 CrossRef CAS; (d) A. Molina-Ontoria, G. Fernández, M. Wielopolski, C. Atienza, L. Sánchez, A. Gouloumis, T. Clark, N. Martin and D. Guldi, J. Am. Chem. Soc., 2009, 131, 12218 CrossRef CAS.
- (a) J. Becher, T. Brimert, J. O. Jeppesen, J. Z. Pedersen, R. Zubarev, T. Bjørnholm, N. Reitzel, T. R. Jensen, K. Kjaer and E. Levillain, Angew. Chem., Int. Ed., 2001, 40, 2497 CrossRef CAS; (b) S. Leroy-Lhez, J. Baffreau, L. Perrin, E. Levillain, M. Allain, M. Blesa and P. Hudhomme, J. Org. Chem., 2005, 70, 6313 CrossRef CAS; (c) C. Jia, S. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem.–Eur. J., 2007, 13, 3804 CrossRef CAS; (d) S. Dolder, S. Liu, F. Derf, M. Sallé, A. Neels and S. Decurtins, Org. Lett., 2007, 9, 3753 CrossRef CAS; (e) K. Qvortrup, A. D. Bond, A. Nielsen, C. J. Mckenzie, K. Kilså and M. B. Nielsen, Chem. Commun., 2008, 1986 RSC; (f) A. Andersson, F. Diederich and M. Nielsen, Org. Biomol. Chem., 2009, 7, 3474 RSC; (g) J. Balandier, M. Chas, P. Dron, S. Goeb, D. Canevet, A. Belyasmine, M. Allain and M. Sallé, J. Org. Chem., 2010, 75, 1589 CrossRef CAS.
- (a) H. Wu, D. Zhang, L. Su, K. Ohkubo, C. Zhang, S. Yin, L. Mao, Z. Shuai, S. Fukuzumi and D. Zhu, J. Am. Chem. Soc., 2007, 129, 6839 CrossRef CAS; (b) H. Wu, D. Zhang, G. Zhang and D. Zhu, J. Org. Chem., 2008, 73, 4271 CrossRef CAS; (c) H. Wu, D. Zhang and D. Zhu, Tetrahedron Lett., 2007, 48, 8951 CrossRef CAS; (d) Y. Zeng, G. Zhang, D. Zhang and D. Zhu, J. Org. Chem., 2009, 74, 4375 CrossRef CAS.
- (a) F. Buchholtz, A. Zelichenok and V. Krongauz, Macromolecules, 1993, 26, 906 CrossRef CAS; (b) M. Lahav, E. Katz, A. Doron, F. Patolsky and I. Willner, J. Am. Chem. Soc., 1999, 121, 862 CrossRef CAS; (c) A. J. Myles and N. R. Branda, J. Am. Chem. Soc., 2001, 123, 177 CrossRef CAS; (d) J. M. Kim, H. Y. Shin, K. H. Park, T. H. Kim, S. Y. Ju, D. K. Han and K. D. Ahn, Macromolecules, 2001, 34, 4391; (e) A. J. Myles, B. Gorodetsky and N. R. Branda, Adv. Mater., 2004, 16, 922 CrossRef CAS; (f) A. J. Myles and N. R. Branda, Tetrahedron Lett., 2000, 41, 3785 CrossRef CAS; (g) R. Born, W. Fischer, D. Heger, B. Tokarczyk and J. Wirz, Photochem. Photobiol. Sci., 2007, 6, 552 RSC.
- The detailed synthetic procedures and characterization data for 1, 2 and 4 are provided in ESI.
- It should be noted that the photoisomerization process of the PNQ unit in dyad 1 is slow compared to that of compound 2 without the TTF unit as shown in Fig. S2, where the variation of the absorbance at 450 nm vs. the UV light irradiation time for dyad 1 and compound 2 is displayed. This is probably due to the photoinduced electron transfer between the TTF and PNQ units in dyad 1, and as a result the excited state of PNQ will be quenched to some extent.
- X. Guo, D. Zhang, H. Zhang, Q. Fan, W. Xu, X. Ai, L. Fan and D. Zhu, Tetrahedron, 2003, 59, 4843 CrossRef CAS.
- (a) P. P. Ashton, V. Balzani, J. Becher, A. Credi, M. C. T. Fyfe, G. Matersteig, S. Menzer, M. B. Nielen, F. M. Raymo, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 3951 CrossRef CAS; (b) H. Spanggaard, J. Prehn, M. B. Nielsen, E. Levillain, M. Allain and J. Becher, J. Am. Chem. Soc., 2000, 122, 9486 CrossRef.
- (a) S. Fukuzumi, N. Nishizawa and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1985, 371 RSC; (b) S. Fukuzumi, K. Hironaka, N. Nishizawa and T. Tanaka, Bull. Chem. Soc. Jpn., 1983, 56, 2220 CAS.
- The other oxygen atoms may also bind metal ion to further stabilize the charge-transfer state.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization data for 1, 2 and 4; other related data. See DOI: 10.1039/c0cc02110e |
| This journal is © The Royal Society of Chemistry 2011 |