Synthesis of C-furanosides from a D-glucal-derived cyclopropane through a ring-expansion/ring-contraction sequence†‡
Russell J.
Hewitt
and
Joanne E.
Harvey
*
School of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington, New Zealand. E-mail: joanne.harvey@vuw.ac.nz; Fax: +64 4 4635237; Tel: +64 4 4635956
First published on 20th September 2010
Abstract
gem-Dibromocyclopropane 1, prepared from tri-O-benzyl-D-glucal, undergoes thermal and silver-promoted ring expansion in the presence of alcohols to give substituted oxepines. With further heating, ring contraction to highly substituted tetrahydrofurans follows. These represent C-furanosides, potentially useful as precursors to C-nucleosides and other carbohydrate mimics.
Carbohydrates are valuable chiral pool reagents1 due to their degree of functionalisation and defined stereochemistries. Cyclopropanation of carbohydrate scaffolds2 enables the preparation of a diverse range of modified sugars, such as C-branched glycosides3,4 and septanoside precursors.4–7Carbohydrate mimics such as these are sought after for, inter alia, their inhibition of carbohydrate-processing enzymes.8 Halogenated cyclopropanes are widely used in synthesis on account of their controllable chemistry.9 Jayaraman and co-workers have demonstrated the utility of 2-oxyglycal-derived gem-dihalocyclopropanes in generating a range of septanosides, including di- and tri-saccharides.6 We have recently shown that gem-dibromocyclopropane 1, derived from D-glucal, provides 2-branched pyranosides 2–4 upon treatment with basic oxygen nucleophiles (Scheme 1).4 Alternatively, silver-promoted ring expansion in the presence of nucleophilic acetate efficiently delivers oxepine 5 in a reasonable yield and as a 3.5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 mixture of C1-stereoisomers.4
∶1 mixture of C1-stereoisomers.4
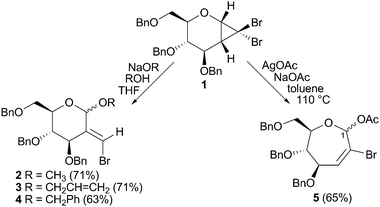 | ||
| Scheme 1 Divergent reactions of D-glucal-derived cyclopropane 1.4 | ||
The pursuit of a range of seven-membered cyclic products related to 5 led us to explore the silver-promoted reactions of cyclopropane 1 with allyl alcohol and methanol. As with the formation of acetate 5,4 a temperature of approximately 100 °C was necessary for efficient cyclopropane ring opening. This is in striking contrast to ring expansions of the lower homologues, bicyclo[3.1.0] systems, which typically proceed at ambient temperature in the presence of silver salts.10 Synthesis of the allyloxy compound 6 was carried out in refluxing allyl alcohol (bp 98 °C), with silver acetate added to promote bromide abstraction and ring opening (Scheme 2). As shown in Table 1, the desired oxepine 6 was isolated as a mixture of C1-epimers,§ along with a small amount of the acetate product 5 (entry 1). To circumvent the formation of 5, use of a non-nucleophilic counterion seemed prudent. However, the yield of the expected product 6 obtained with silver nitrate (entry 2) was no better than with silver acetate, despite the lack of a competing nucleophilic silver counter-ion. It was postulated that the nitric acid by-product is detrimental to the acetal functionality within 6, and this was borne out by the subsequent experiment in which the reaction was refluxed for an extended time (entry 3). After 3 days, none of the expected oxepine 6 was isolated; instead, three compounds bearing highly functionalised tetrahydrofuran rings were obtained in a combined yield of 51%. One was assigned the structure 7, which contains a brominated enol ether side-chain. The remaining two compounds appeared to be diastereomers represented by structure 8. These assignments were made on the basis of the obtained 1D and 2D NMR data,¶|| and were supported by mass spectrometry.
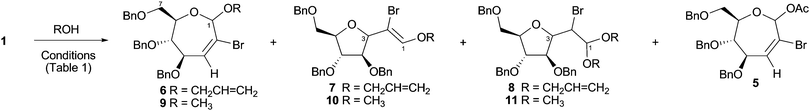 | ||
| Scheme 2 Formation of oxepines and/or substituted tetrahydrofurans from cyclopropane 1. | ||
| Entry | ROH | Additive | Temperature | Time | Yielda | ||||
|---|---|---|---|---|---|---|---|---|---|
| 6 or 9b | 7 or 10 | 8 or 11b | 5 | 1 | |||||
|
a Yields quoted are isolated yields, with the exception of compounds 1, 10 and 11 in entry 8, in which the quantity of compound 1 was estimated from the 1H NMR spectrum of the crude reaction mixture, and 10 and 11 were detected by TLC.
b Ratios quoted are calculated from the yields of the separated stereoisomers, and are shown as the isomer with higher TLC mobility followed by the more polar isomer. For 6 and 9, the isomers have been assigned such that the ratios give β-anomer∶α-anomer.
c This ratio is of the isolated but unseparated isomers of 8.
|
|||||||||
| 1 | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OH CHCH2OH |
AgOAc | 98 °C | 27 h | 51%, 1.1∶1 |
0% | 0% | 4% | 13% |
| 2 | CH2CHCH2OH |
AgNO3 | 98 °C | 29 h | 50%, 1∶1.8 |
0% | 0% | 0% | 16% |
| 3 | CH2CHCH2OH |
AgNO3 | 98 °C | 3 d | 0% | 15% | 36%, 1∶2.5 |
0% | 0% |
| 4 | CH2CHCH2OH |
AgOCOCF3 | 98 °C | 3 d | 22%, 1∶1.8 |
22% | 8%, ca. 1∶10c |
0% | 5% |
| 5 | CH2CHCH2OH |
None | 98 °C | 2 d | 0% | 12% | 45%, 1∶1.6 |
0% | 0% |
| 6 | CH3OH | AgOAc | 65 °C | 5 d | 15%, 1∶2.8 |
0% | 0% | 0% | 61% |
| 7 | CH3OH | None | 130 °C (MW) | 1 h | 0% | 13% | 61%, 1∶3 |
0% | 4% |
| 8 | CH3OH | AgOAc | 100 °C (MW) | 10 min | 4%, 3∶1 |
Trace | Trace | 0% | ca. 70% |
Use of silver trifluoroacetate as the promoter gave a mixture of oxepine 6, tetrahydrofurans 7 and 8 after 3 days reaction in allyl alcohol (entry 4). Finally, an attempt to ring expand cyclopropane 1 in the absence of silver salts produced, after 2 days of refluxing in allyl alcohol, the tetrahydrofuran products 7 and 8 in a combined yield of 57% (entry 5).
In contrast to the reactivity with allyl alcohol, silver-mediated reactions in methanol were sluggish and low yielding. For instance, the silver acetate reaction resulted in 15% yield of oxepines 9 alongside 61% recovered starting material 1 after 5 days (entry 6).4 This is because the requirement for high temperature (ca. 100 °C) to promote the cyclopropane ring expansion in this system is not met by methanol (bp 65 °C). Thereafter, subjection of cyclopropane 1 to thermal ring expansion in methanol was performed in a sealed tube under microwave conditions14 (130 °C, ca. 8 bar) for 1 hour. The enol ether 10 and acetal 11 (the latter as a mixture of two stereoisomers) were isolated in a combined yield of 74%, and no oxepine was observed (entry 7). When the microwave reactions in methanol were halted prematurely (e.g. after only 10 minutes), the crude reaction mixtures contained oxepines 9 as well as traces of C-furanosides 10 and 11 (entry 8).
It appears that the C-furanosyl products are formed via the oxepines upon heating of the cyclopropane ring expansion reaction for prolonged times. In confirmation, heating a solution of the major isomer of oxepine 6 in allyl alcohol at reflux in the presence of silver nitrate gave, after one day, the tetrahydrofurans 7 and 8, with only a trace of the oxepine 6 remaining in the crude reaction mixture (according to 1H NMR spectroscopy). Similarly, reaction of oxepine 6 (as a mixture of epimers) with allyl alcohol in the absence of silver salts led to formation of the acetal isomers 8.
A likely mechanism for the formation of the C-furanoside products from cyclopropane 1 is shown in Scheme 3. Cyclopropyl ring opening is thermally allowed and is promoted by silver(I) salts; attack on the intermediate 12 by an alcohol provides, after deprotonation, the oxepine isomers 13. Acid-promoted ring opening of the oxepine acetal and attack by the ring oxygen at C3 of the highly activated, conjugated alkene in intermediate 14 would lead to the bromoenol ether 15. The observation of only a single isomer of the isolated enol ethers 7 and 10 is consistent with electron delocalisation in oxonium intermediate 14 and preferred attack of the hydroxyl group from one face of the alkene. The bromoenol ether of 15 is highly reactive towards electrophilic addition, and is converted into the acetal 16. The mechanistic processes encompassing transformation of oxepine 13 into 15 and 16 are catalysed by acid and are related to those proposed by Skattebøl in a less substituted system.15 It is noteworthy that the reactions in which these tetrahydrofuran products dominate are those in which a stoichoimetric amount of hydrogen bromide or nitric acid is released upon cyclopropane opening and formation of the oxepines (viz.Table 1, entries 3, 5, 7).
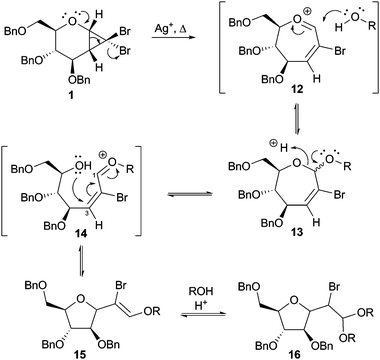 | ||
| Scheme 3 Proposed mechanism of formation of tetrahydrofuran products from cyclopropane 1. | ||
The products obtained are highly functionalised, enantiomerically pure tetrahydrofurans.16 They are closely related to intermediates used previously in the formation of ulosonic acids17 and C-nucleosides.18,19
In conclusion, D-glucal-derived cyclopropane 1 undergoes silver-mediated or thermal ring expansion in alcohol solvents to provide complex oxepines. Further reaction leads to ring contraction and the formation of a series of highly substituted tetrahydrofuran derivatives (7 and 8, 10 and 11); these represent new C-glycosides with potential for elaboration to C-nucleosides and glycosidase inhibitors.
The authors gratefully acknowledge the Cancer Society of New Zealand for a PhD scholarship (RJH). We extend our thanks to Dr Brendan Burkett for advice and assistance with the microwave experiments.
Notes and references
- G. Casiragha, F. Zanardi, G. Rassu and P. Spanu, Chem. Rev., 1995, 95, 1677–1716 CrossRef CAS.
- (a) B. Fraser–Reid and J. C. López, Curr. Org. Chem., 2009, 13, 532–553 CrossRef CAS; (b) J. S. Brimacombe, M. E. Evans, E. J. Forbes, A. B. Foster and J. M. Webber, Carbohydr. Res., 1967, 4, 239–243 CrossRef CAS.
- (a) P. R. Sridhar, P. V. Kumar, K. Seshadri and R. Satyavathi, Chem.–Eur. J., 2009, 15, 7526–7529 CrossRef CAS; (b) D. W. Gammon, H. H. Kinfe, D. E. De Vos, P. A. Jacobs and B. F. Sels, J. Carbohydr. Chem., 2007, 26, 141–157 CrossRef CAS; (c) H. Shao, S. Ekthawatchai, C.-S. Chen, S.-H. Wu and W. Zou, J. Org. Chem., 2005, 70, 4726–4734 CrossRef CAS; (d) C. V. Ramana, R. Murali and M. Nagarajan, J. Org. Chem., 1997, 62, 7694–7703 CrossRef CAS; (e) K. J. Henry Jr. and B. Fraser-Reid, Tetrahedron Lett., 1995, 36, 8901–8904 CrossRef CAS.
- R. J. Hewitt and J. E. Harvey, J. Org. Chem., 2010, 75, 955–958 CrossRef CAS.
- (a) R. Batchelor and J. O. Hoberg, Tetrahedron Lett., 2003, 44, 9043–9045 CrossRef CAS; (b) J. O. Hoberg, J. Org. Chem., 1997, 62, 6615–6618 CrossRef CAS; (c) J. O. Hoberg and J. J. Bozell, Tetrahedron Lett., 1995, 38, 6831–6834.
- (a) N. V. Ganesh, S. Raghothama, R. Sonti and N. Jayaraman, J. Org. Chem., 2010, 75, 215–218 CrossRef CAS; (b) N. V. Ganesh and N. Jayaraman, J. Org. Chem., 2009, 74, 739–746 CrossRef CAS.
- (a) R. Batchelor, J. E. Harvey, P. T. Northcote, P. Teesdale-Spittle and J. O. Hoberg, J. Org. Chem., 2009, 74, 7627–7632 CrossRef CAS; (b) M. W. Peczuh and N. L. Snyder, Tetrahedron Lett., 2003, 44, 4057–4061 CrossRef CAS; (c) M. P. DeMatteo, N. L. Snyder, M. Morton, D. M. Baldisseri, C. M. Hadad and M. W. Peczuh, J. Org. Chem., 2005, 70, 24–38 CrossRef CAS; (d) J. C. Y. Wong, P. Lacombe and C. F. Sturino, Tetrahedron Lett., 1999, 40, 8751–8754 CrossRef CAS.
- (a) F. Nicotra, L. Cipolla, B. La Ferla, C. Airoldi, C. Zona, A. Orsato, N. Shaikh and L. Russo, J. Biotechnol., 2009, 144, 234–241 CrossRef CAS; (b) B. G. Winchester, Tetrahedron: Asymmetry, 2009, 20, 645–651 CrossRef CAS; (c) D. Sabatino and M. J. Damha, J. Am. Chem. Soc., 2007, 129, 8259–8270 CrossRef CAS; (d) J. Shao, B. Zhou, B. Chu and Y. Yen, Curr. Cancer Drug Targets, 2006, 6, 409–431 Search PubMed; (e) A. Tauss, A. J. Steiner, A. E. Stütz, C. A. Tarling, S. G. Withers and T. M. Wrodnigg, Tetrahedron: Asymmetry, 2006, 17, 234–239 CrossRef CAS; (f) S. Castro, M. Duff, N. L. Snyder, M. Morton, C. V. Kumar and M. W. Peczuh, Org. Biomol. Chem., 2005, 3, 3869–3872 RSC.
- (a) M. G. Banwell, Pure Appl. Chem., 2008, 80, 669–679 CrossRef CAS; (b) B. Halton and J. Harvey, Synlett, 2006, 1975–2000 CAS; (c) M. Fedoryński, Chem. Rev., 2003, 103, 1099–1132 CrossRef CAS.
- It has been noted that bicyclo[3.1.0] systems undergo ring expansionca. 200 times faster than bicyclo[4.1.0] and monocyclic gem-dihalocyclopropanes: see P. S. Skell and S. R. Sandler, J. Am. Chem. Soc., 1958, 80, 2024–2025 Search PubMed.
- H. Takayama, T. Koike, N. Aimi and S.-i. Sakai, J. Org. Chem., 1992, 57, 2173–2176 CrossRef CAS.
- S. Y.-K. Tam, R. S. Klein, F. G. de las Heras and J. J. Fox, J. Org. Chem., 1979, 44, 4854–4862 CrossRef CAS.
- H. Ohrui, G. H. Jones, J. G. Moffatt, M. L. Maddox, A. T. Christensen and S. K. Byram, J. Am. Chem. Soc., 1975, 97, 4602–4613 CrossRef CAS.
- A. De la Hoz, A. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- L. Skattebøl, J. Org. Chem., 1970, 35, 3200–3201 CrossRef.
- F. Freeman and K. D. Robarge, Tetrahedron Lett., 1985, 26, 1943–1946 CrossRef CAS.
- (a) R. Csuk, U. Franke, Z. Hu and C. Krieger, Tetrahedron, 2003, 59, 7887–7895 CrossRef CAS; (b) G. V. M. Sharma, Rakesh, A. Subhash Chander, V. Goverdhan Reddy, M. H. V. Ramana Rao and A. C. Kunwar, Tetrahedron: Asymmetry, 2003, 14, 2991–3004 CrossRef CAS.
- (a) P. J. Dudfield, V.-D. Le, S. D. Lindell and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1999, 2929–2936 RSC; (b) M. C. Clingerman and J. A. Secrist III, J. Org. Chem., 1983, 48, 3141–3145 CrossRef CAS.
- M. S. Pino Gonzalez, R. M. Dominguez Aciego and F. J. Lopez Herrera, Tetrahedron, 1988, 44, 3715–3726 CrossRef.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all new compounds. See DOI: 10.1039/c0cc02244f |
| § In the crude mixtures obtained from all the silver-mediated reactions of 1 in allyl alcohol, the ratio of allyloxy-substituted oxepines 6 was ca. 1∶1.4, as judged by 1H NMR integration. The major allyloxy-substituted oxepine epimer has been assigned as the 1S-isomer (the α-anomer, using carbohydrate nomenclature) on the basis of a nOe enhancement of the H-7 signal upon irradiation of H-1. Similar ratios were observed in reactions with methanol, and the 1H and 13C NMR shifts of the methoxy compounds correlated very closely with the allyloxy derivatives, leading to assignment of the major component of 9 as the α-anomer. However, the isolated ratios varied between experiments. For instance, in Table 1, entry 1, the isomer that was isolated in a greater quantity was actually the minor isomer. This indicates that erosion of the true yield of the major isomer occurred during column chromatography in our efforts to obtain pure material. |
| ¶ The enol ether moiety of 7 was evident in the NMR spectra by virtue of the alkene proton signal at δ 6.69, which was directly connected (HSQC) to a carbon at δ 146. This position demonstrated a long-range coupling (HMBC) with an allyloxy group, indicating the two groups might comprise an enol ether moiety. It also showed HMBC correlations with an unprotonated carbon resonating at δ 101.6, which was assigned as the adjacent olefinic carbon at C-2, and an oxymethine at δ 82.4 (C-3). No HMBC correlation was observed between positions 3 and 6; however, the signals were fully consistent with the tetrahydrofuran ring structure of 7. Assignment of the enol ether stereochemistry came from treatment of the bromoenol ether 7 with n-butyllithium, to effect bromine-for-lithium exchange, and a subsequent water quench. While the product was not isolated, the singlet corresponding to H-1 in the 1H NMR spectrum was now observed at δ 6.53 as a doublet with a coupling constant of 12.5 Hz, indicative of an enol ether with an E-configuration.11 Tentative assignment of the stereochemistry of enol ether 7 as 3R (β-pseudo-anomer) has been made on the basis of the 1H NMR shift of H-3 (δ 4.37) in comparison with related compounds.12 The structural and stereochemical determination of the methyl variant 10 was made similarly. |
| || The two partially separable isomers of 8 were obtained in a 1∶2.5 ratio. Both contained an acetal signal in their 13C NMR spectra, at δ 101.9 and δ 100.2, respectively, which were directly connected (HSQC) to protons at δ 4.80 and δ 4.44, respectively. The acetal position in the major isomer showed HMBC cross-peaks to two allyl groups and a methine (δHca. 4.28, δC 52.3). The chemical shifts and correlation pattern indicated that this latter centre could be a bromomethine functional group. This system displays an HMBC correlation with an oxymethine at C-3 that is part of the tetrahydrofuran ring. The minor isomer displayed similar spectroscopic properties and was assigned as a diastereoisomer of the same structure. It seems likely that the two observed compounds of structure 8 are epimers at C-2 (the bromomethine) because the stereochemistry at C-3 would not be expected to interchange in the formation of the acetal isomers from enol ether 7. The same arguments are applicable to dimethyl acetal 11. The fact that the 13C chemical shifts of the C-3 position in all four compounds are so consistent (allyl isomers: δ 81.7 and 81.0, methyl isomers: δ 79.9 and 81.1) lends credence to these assignments. Furthermore, the magnitudes of these chemical shifts compare favourably with similar compounds that were assigned the β-pseudo-anomeric configuration, while being significantly downfield of those assigned the opposite configuration.13 |
| This journal is © The Royal Society of Chemistry 2011 |