Double addition of H2 to transition metal–borane complexes: a ‘hydride shuttle’ process between boron and transition metal centres†‡
Nikolaos
Tsoureas
,
Yu-Ying
Kuo
,
Mairi F.
Haddow
and
Gareth R.
Owen§
*
The School of Chemistry, University of Bristol, Bristol, UK. E-mail: gareth.owen@bristol.ac.uk; Fax: +44 (0)117 925 1295; Tel: +44 (0)117 928 7652
First published on 11th October 2010
Abstract
The addition of H2 across a transition metal–borane bond is reported for the first time providing a mechanism for recharging borane functional groups to borohydride.
The synthesis of the first authenticated transition metal–borane complexes, the so-called ‘metallaboratranes’,1 in 1999 enriched the fields of organometallic, coordination and transition metal–boron chemistry.2 Since this time, a wide range of diverse transition metal–borane complexes have been prepared. Such complexes are of particular interest due to the sigma acceptor (Z-class) interaction between the boron and metal centre.2,3 Over the course of the past few years we have carried out a number of investigations aimed at utilizing this class of compounds as ‘hydride shuttles’; probing a ‘BH cooperation effect’ involving reversible migration of a hydride species between boron and transition metal centres.4 Control over hydride migration could have wide ranging scope, however, little is known regarding such reactivity. In particular, little is known concerning addition reactions across these bonds. Parkin et al. have previously reported 1,2-addition reactions of halogens to this bond in a number of complexes.5 While contrasting evidence was found by Hill et al. who subjected a platinum–borane complex to oxidants such as Cl2, Br2, I2 and CH3I and found retention of the Pt–B bond.6 The reactivity of transition metal–borane bonds remains elusive and to date there have been no reports of its reactivity towards H2. A clear insight of such reactivity could have potential applications in processes such as hydrogen storage technologies, catalysis and contemporary organometallic transformations.
Sabo-Etienne et al. have recently reported a ruthenium complex which exhibits reversible hydrogen release.7 In this complex, a reversible transformation was observed between borylene and bis(σ-borane) functionalities. Weller et al. have also shown reversible hydrogen addition to a rhodium complex containing a σ-bound carborane.8 Herein, we report for the first time, the successful demonstration of H2 addition to a transition metal–borane complex and its utilization in the catalytic hydrogenation of olefins.
We have previously reported the reactivity of [Rh{κ3-NNH-HB(azaindolyl)3}(NBD)] (1) (where NBD = η2-norbornadiene) to form the rhodium–borane complex, [Rh{κ4-NNBN-B(azaindolyl)3}(nortricyclyl)] (2) (Scheme 1).4b In this transformation, the hydride migrates from the boron atom into the former norbornadiene ligand followed by a double bond rearrangement to form the nortricyclyl moiety. The reactivity of complex 2 with trialkylphosphines was explored further. Complex 2 readily reacts with a slight excess of PMe3 to give complex 3 (Scheme 2; see ESI‡ for details of synthesis and structural characterisation). When the reaction was repeated using bulkier trialkylphosphines [such as PtBu3, PCyp3 (Cyp = cyclopentyl), PCy3] no significant conversion of the starting material was observed even at elevated temperatures or prolonged periods of time.
![Reaction of [Rh{κ3-NNH-HB(azaindolyl)3}(NBD)] (1) to form [Rh{κ4-NNBN-B(azaindolyl)3}(nortricyclyl)] (2).](/image/article/2011/CC/c0cc02245d/c0cc02245d-s1.gif) | ||
| Scheme 1 Reaction of [Rh{κ3-NNH-HB(azaindolyl)3}(NBD)] (1) to form [Rh{κ4-NNBN-B(azaindolyl)3}(nortricyclyl)] (2). | ||
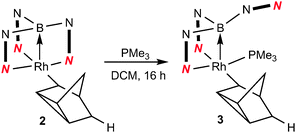 | ||
| Scheme 2 Addition of trialkylphosphines to complex 2. | ||
Complex 2 was dissolved in toluene-d8 and one equivalent of tricyclopentylphosphine was added. The mixture was placed under 2.5 bar of H2 at 85 °C (Scheme 3). After 16 h, the 11B{1H} spectrum showed the conversion of complex 2 into two new species; a major species (ca. 95%, as determined by relative integration) as a singlet at −0.6 ppm (4) and a small quantity of a second species with a signal centred at 3.5 ppm (ca. 5%). The identity of the minor species is described below (complex 6). Complex 4 was isolated as a spectroscopically and analytically pure microcrystalline solid (see ESI‡). The spectroscopic data were consistent with the formation of [Rh{κ3-NNH-HB(azaindolyl)3}(H)2(PCyp3)] (4) (Scheme 3). The 11B{1H} NMR spectrum (s, Δν1/2 = 20 Hz) confirmed that the rhodium–boron bond have been lost and the corresponding boron–coupled experiment confirmed the formation of a new borohydride functionality (1JBH = 75 Hz). The chemical shift and coupling constant found in 4 are similar to those found for complex 1 (cf. −1.6 ppm; 1JBH = 78 Hz).4b The proton NMR spectrum was notable for the presence of three hydridicprotons; one at −4.25 ppm (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1∶1∶1 quartet, 1JBH = 73 Hz)¶ integrating for one hydrogen atom (BH) and two overlapping signals centred at −16.30 ppm as broad multiplet signals integrating for two hydrogen atoms.
∶1∶1∶1 quartet, 1JBH = 73 Hz)¶ integrating for one hydrogen atom (BH) and two overlapping signals centred at −16.30 ppm as broad multiplet signals integrating for two hydrogen atoms.
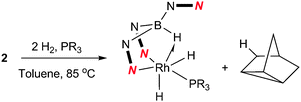 | ||
| Scheme 3 Reaction of 2 with H2 and trialkylphosphines to give 4 (R = Cyp) and 5 (R = tBu). | ||
The analogous reaction was carried out with PtBu3 to form [Rh{κ3-NNH-HB(azaindolyl)3}(H)2(PtBu3)] (5). In this case, a larger quantity (up to 25%) of a second species was observed in the reaction mixtures. This species was the same as that found in the reaction involving PCyp3 (i.e. complex 6). The identity of 5 was further confirmed by structural characterisation (Fig. 1). The molecular structure revealed an octahedral rhodium(III) centre bound by Tai [Tai = HB(azaindolyl)3], coordinating with a κ3-NNH coordination mode, one hydride ligand located cis to the BH group and one trans to the BH group. A PtBu3 ligand is additionally coordinated cis to the BH group. The presence of two terminal hydride ligands was confirmed by the 1H NMR spectrum which revealed two distinct chemical environments with the corresponding signals at −17.2 ppm and −16.92 ppm.
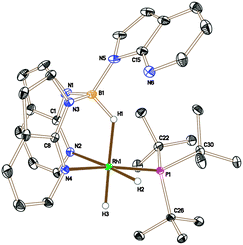 | ||
| Fig. 1 Crystal structure of 5 (solvent and hydrogen atoms, with the exception of H1, H2 and H3, have been removed for clarity, ellipsoids drawn at 50% probability level) Selected bond lengths (Å): Rh1–N4 2.1159(7), Rh1–N2 2.1896(8), Rh1–P1 2.2928(3), N1–B1 1.5336(13), N3–B1 1.5386(12), N5–B1 1.5323(14).††|| | ||
The higher proportion of the unknown species 6 in the reaction involving the bulkier phosphine PtBu3 suggested that it might not contain a phosphine ligand. Therefore, in a control experiment, 2 was placed under the same conditions as those shown in Scheme 3 in the absence of a phosphine ligand. The 11B{1H} NMR spectrum of the reaction mixture indicated the complete conversion of 2 and the formation of 6 as the major species, although some decomposition was also apparent in the mixture. Isolation and structural characterisation of 6 revealed its identity as the unexpected product [(Rh{κ3-NNB-B(azaindolyl)3})2(μ-H)(μ-azaindolyl)] (Fig. 2). Single crystals of 6 were obtained by allowing a diethyl ether solution to stand at −30 °C overnight. The molecular structure of 6 consists of a dimeric species where the rhodium centres adopt pseudo square based pyramidal geometries with coordination spheres comprising of two nitrogen atoms and the boron atom of the former Tai ligand (with a κ3-NNB coordination mode). An azaindolyl unit bridges the two rhodium centres via its two nitrogen atoms** and an additional hydride ligand bridges the two metal centres.†† The 1H NMR spectrum of 6 gave some signals which were broad at ambient temperatures indicating fluxional behaviour within the complex. At −60 °C, the signals sharpened revealing several azaindole ring environments with a total integration for 35 protons. A signal integrating for one proton was also observed at −22.4 ppm (t, 1JRhH = 23.5 Hz) confirming the presence of the bridging hydride. The hydride signal remained as a broad triplet at lower temperatures even though two boron chemical environments were observed in the 11B NMR spectrum between the temperatures −60 °C to 80 °C.
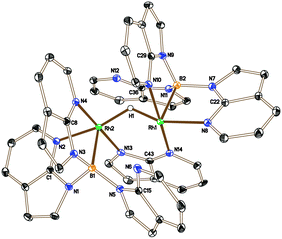 | ||
| Fig. 2 Crystal structure of 6 (solvent and hydrogen atoms, with the exception of H1, have been removed for clarity, ellipsoids drawn at 50% probability level). Selected bond lengths (Å): Rh1–N14 2.027(3), Rh1–N2 2.100(3), Rh1–N4 2.024(3), Rh1–B1 2.171(4), Rh1–Rh2 2.8803(4), Rh2–N13 2.046(3), Rh2–N8 2.095(3), Rh2–N10 2.095(3), Rh2–B1 2.139(5).††||‡‡ | ||
The formation of 6 suggests that a highly-reactive unsaturated rhodium species, such as [(Rh{κ3-NNB-B(azaindolyl)3}(μ-H)]2 (A*) (Scheme 4), is formed during the hydrogenation reaction pathway. This species can either be captured by a phosphine ligand to form complex 4 or 5 (following subsequent reaction with H2) or undergo decomposition in the absence of a trialkylphosphine ligand. The presence of a bridging azaindolyl unit in 6 suggests that the unsaturated species (A*) reacts with free azaindole which is generated by an unknown decomposition pathway involving B–N bond fission (Scheme 4). Indeed repeating the same reaction in the presence of 0.5 equivalents of 7-azaindole resulted in complex 6 as the only boron-containing species observed in the 11B NMR spectrum (see ESI‡). The involvement of such an intermediate was also confirmed by the inability of 3 to react with H2 under the same conditions.
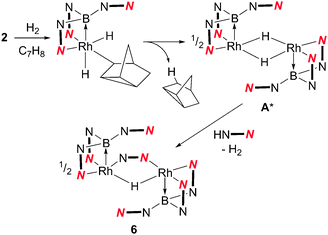 | ||
| Scheme 4 Reaction of 2 with H2 (A* is a proposed intermediate, see text for details). | ||
The complexes presented herein represent the first examples exhibiting reactivity of a transition metal–borane bond with H2. The conversion of complex 1 to complexes 4 and 5 involves a stepwise transfer of hydrogen atoms. A suggested mechanism is shown in Scheme 5, in which the first hydrogen originates from boron. Addition of hydrogen gas to the system allows for elimination of tricyclo[2.2.1.02,6]heptane and the boron atom is recharged with a hydride ligand.
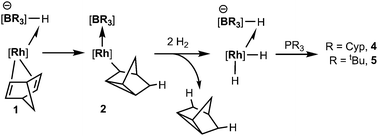 | ||
| Scheme 5 Suggested pathway involving the transformation of 1 to complexes 4 and 5 (some substituents at boron and rhodium have been removed for clarity). | ||
This reactivity prompted us to investigate the potential of our complexes as hydrogenation catalysts (Table 1). Preliminary investigations reveal that under 1 mol% of 2, the substrates styrene and cyclooctene are fully converted to ethyl benzene and cyclooctane, respectively, at 2.5 bar H2 at 85 °C over a period of 18 h. At lower catalyst loading (0.1 mol%), these substrates were reduced to their corresponding alkanes with 85% and 43% conversions. When the phosphine complex 4 was employed as the catalyst, these conversions were reduced to 50% and 7%. This result suggests that the phosphine ligand is eliminated from the complex during the catalytic cycle. Further investigations are currently being carried out to explore the borane recharge mechanism and the role of borane and phosphine ligands within the catalytic cycle.
| Complex | Cat. loading (mol%) | Substrate | Conversiond (%) |
|---|---|---|---|
| a 2.5 bar H2, 85 °C. b 2 mmol of olefin, 0.02 mmol of 2 or 4, C6D6 (2 mL). c 7 mmol of olefin, 0.007 mmol of 2 or 4, C6D6 (1 mL). d Conversion measured by NMR integration relative to internal standard after 18 h. | |||
| 2 | 1.0b | Styrene | >99 |
| 2 | 0.1c | Styrene | 85 |
| 4 | 0.1c | Styrene | 50 |
| 2 | 1.0b | Cyclooctene | >99 |
| 2 | 0.1c | Cyclooctene | 43 |
| 4 | 0.1c | Cyclooctene | 7 |
The authors would like to thank the Royal Society (G.R.O.) and Leverhulme Trust (N.T.) for funding, Johnson Matthey for the loan of the rhodium salts, Dr C. P. Butts for his help with the NMR studies and the EPSRC National Crystallography Service (University of Southampton) for the data collection for complex 6.
Notes and references
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759 CrossRef CAS
.
-
(a) I. Kuzu, I. Krummenacher, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836 RSC
-
(a) R. B. King, Adv. Chem., 1967, 62, 203 Search PubMed
-
(a) G. R. Owen, N. Tsoureas, A. Hamilton and A. G. Orpen, Dalton Trans., 2008, 6039 RSC
-
(a) K. Pang, J. M. Tanski and G. Parkin, Chem. Commun., 2008, 1008 RSC
- I. R. Crossely, A. F. Hill and A. C. Willis, Organometallics, 2008, 27, 312 CrossRef CAS
-
(a) G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878 CrossRef CAS
- E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Dalton Trans., 2007, 4829 RSC
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details outlining the synthesis and characterisation of 3–6 and further details and structural data. CCDC 783205–783209. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02245d |
| § Royal Society Dorothy Hodgkin Research Fellow. |
| ¶ Further coupling is apparent in this signal, however the signal was too broad to resolve the coupling constants. |
|| Crystal data for complex 5: C33H45BN6PRh·0.5(C5H12), M = 706.51, monoclinic, a = 11.6549(8) Å, b = 12.9893(9) Å, c = 22.6123(15) Å, α = 90.00°, β = 90.483(3)°, γ = 90.00°, V = 3423.1(4) Å3, T = 100(2) K, space groupP21/c, Z = 4, 109624 reflections measured, 21767 independent reflections (Rint = 0.0298). The final R1 values were 0.0298 (I > 2σ(I)). The final wR(F2) values were 0.0743 (I > 2σ(I)). The final R1 values were 0.0402 (all data). The final wR(F2) values were 0.0807 (all data). Crystal data for complex 6: C57H56B2N14O2Rh2, M = 1196.60, triclinic, a = 11.3780(3) Å, b = 11.9021(3) Å, c = 20.7067(6) Å, α = 74.070(2)°, β = 78.342(2)°, γ = 80.084(2)°, V = 2620.47(12) Å3, T = 100(2) K, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 53963 reflections measured, 12055 independent reflections (Rint = 0.0941). The final R1 values were 0.0504 (I > 2σ(I)). The final wR(F2) values were 0.1015 (I > 2σ(I)). The final R1 values were 0.0972 (all data). The final wR(F2) values were 0.1164 (all data). , Z = 2, 53963 reflections measured, 12055 independent reflections (Rint = 0.0941). The final R1 values were 0.0504 (I > 2σ(I)). The final wR(F2) values were 0.1015 (I > 2σ(I)). The final R1 values were 0.0972 (all data). The final wR(F2) values were 0.1164 (all data). |
| ** The structure reveals that the bridging azaindolyl unit occupies the two possible bridging orientations with equal disorder. |
| †† Full details regarding the collection parameters and refinement for complexes 3, 5 and 6 (CCDC 783205–783209) are presented in the ESI.‡ |
| ‡‡ Complex 6 was also crystallised as a second polymorph. |
| This journal is © The Royal Society of Chemistry 2011 |