The art of stacking: structural folding and self-assembly of branched π-conjugation assisted by O–H⋯O and C–H⋯F hydrogen bonds†‡
Ho Yong
Lee
,
András
Olasz
,
Maren
Pink
,
Hyunsoo
Park
and
Dongwhan
Lee
*
Department of Chemistry, Indiana University, 800 East Kirkwood Avenue, Bloomington, IN 47405, USA. E-mail: dongwhan@indiana.edu; Fax: +1 812-855-8300; Tel: +1 812-855-9364
First published on 11th October 2010
Abstract
An intimate interplay of O–H⋯O/C–H⋯F hydrogen bonds and π–π stacking interactions allows a phenyleneethynylene-based dendritic molecule to fold and self-assemble into two distinctively different molecular crystals as pseudopolymorphs.
Polymorphism describes the ability of molecules to adopt more than one structure in the solid state.1 When polymorphs occur with variations in solvent contents in the unit cell, the term pseudopolymorphism is used.2 A practical importance of this phenomenon has been recognised in the field of pharmaceuticals since (pseudo)polymorphs often display different physicochemical properties.1a,1c,3,4 In this communication, we report pseudopolymorphism of a phenyleneethynylene-based C3-symmetric π-conjugated molecule. Structure analysis on two distinctively different molecular crystals revealed an intimate interplay of strong O–H⋯O and weak C–H⋯F hydrogen bonds in structural folding and solid-state self-assembly (Scheme 1), a detailed understanding of which was also aided by theoretical studies.
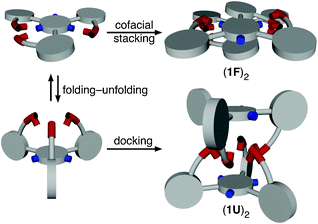 | ||
| Scheme 1 Conformational switching to achieve shape complementarity for self-association. | ||
Hydrogen bonding is one prominent type of non-covalent interaction, the directional nature of which is proven to play a critical role in the spontaneous structural ordering of many chemical architectures.5 While strong hydrogen bonds are typically constructed using N–H⋯O, O–H⋯O, or O–H⋯N motifs, weak hydrogen bonds involving C–H⋯O, C–H⋯N, or C–H⋯π contacts have recently been recognised as an important class of non-covalent interactions.6 Recent examples of linear foldamers and heterodimers that are stabilised by N–H⋯F hydrogen bonding support the notion that organofluorine fragments can also be utilised as a hydrogen bonding acceptor (HBA) for the assembly of higher-order structures.7 Undoubtedly, the charged F− anion is a strong HBA,8 but the ability of covalently bound fluorine atom, as present in the C–F group, to function as an effective HBA still remains controversial.9–12 Statistical analysis on crystallographically determined structures suggests, however, that C–H⋯F interactions could be as important as C–H⋯O or C–H⋯N hydrogen bonds.13
Our on-going interest in the conformational switching of branched π-conjugated structures14 prompted the design and synthesis of a new phenyleneethynylene-based C3-symmetric molecule 1 having a fluorinated molecular core (Scheme 2).‡ The final round of cross-coupling between 4 and 1,3,5-trifluoro-2,4,6-triiodobenzene resulted in a mixture of the desired product 1, along with partially coupled byproducts and the homocoupling product of 4. A simple layering of hexanes over a CHCl3 solution of this material, however, induced selective crystallization of 1, which was isolated and fully characterised (Fig. S1).‡
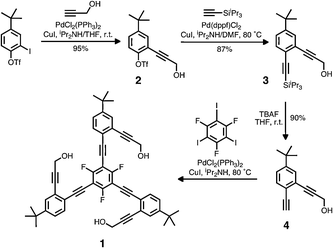 | ||
| Scheme 2 Synthetic route to 1. | ||
Self-association of 1 in solution was subsequently investigated by concentration-dependent 1H NMR studies in CDCl3 at T = 298 K. A systematic downfield shift of the propargylic O–H resonance from 2.52 to 2.73 ppm with increasing concentration within the range of 10 to 40 mM could be fitted with K = 4.1 (±0.5) M−1 using the EK model.15 This observation implicated the involvement of O–H⋯O hydrogen bonding in the self-association process, which should become more favorable with decreasing polarity in the CHCl3/hexane binary solvent mixture used in crystallization.
Intriguingly, two different kinds of single crystals of 1 were obtained depending on the method of crystallization.§ Specifically, colorless blocks of the completely “unfolded” conformer 1U (Fig. 1a) were obtained by slow diffusion of hexane into a CHCl3 solution of 1.16 On the other hand, an instant mixing of a CHCl3 solution of 1 with hexane or benzene produced colorless needles of the “folded” conformer 1F (Fig. 1b),16 which was also characterised by single crystal X-ray crystallography. As shown in Fig. 1 and S2 (ESI‡), the fluorinated benzene core and peripheral aryl groups in 1U are disposed in an essentially perpendicular manner with dihedral angles of τ = 79.3(2) and 83.4(7)°.17,18 For the other conformer 1F, however, two of the three ethynyl-extended “wingtip” O–H groups engage in an intramolecular O–H⋯O hydrogen bond (O⋯O = 2.731(4) Å), which, along with C–H⋯F contact to the fluorinated core (C⋯F = 3.209(5) Å), effectively flattens the entire structure with relatively small τ values of 9.0(2), 14.1(2), and 35.5(1)°.18
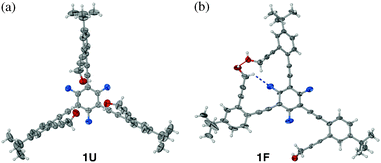 | ||
| Fig. 1 X-Ray structures of 1U (a) and 1F (b) with thermal ellipsoids at 50% probability, where O is red and F is blue. The O–H⋯O and C–H⋯F interactions are represented by red and blue dotted lines, respectively. | ||
A close inspection of the intermolecular packing in the lattice provided an initial clue to the origin of conformational isomerism of 1. Instead of making intramolecular O–H⋯O bond as in 1F (Fig. 1b), 1U uses strong intermolecular O–H⋯O hydrogen bonds (O⋯O = 2.689(5) Å) to form a discrete dimeric structure (1U)2. As shown in Fig. 2 and S3 (ESI‡), the two 1U units comprising the dimer maximise their shape complementarity by adopting an essentially “staggered” orientation, in which two C3-symmetric molecules are rotated by 60 degrees with respect to each other to achieve a tight docking.
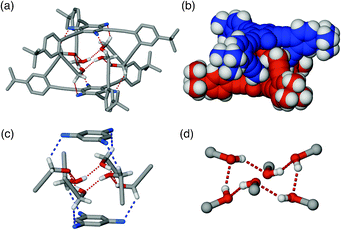 | ||
| Fig. 2 Capped stick (a) and space-filling (b) representations of the solid-state structure of dimeric (1U)2 generated using X-ray coordinates. The O–H⋯O and C–H⋯F interactions are represented by dotted lines, the close-up views of which are provided in partial structures (c) and (d). | ||
An interdigitated arrangement of phenyleneethynylene fragments in the dimeric (1U)2 thus provides a well-shielded hydrophobic cavity that encapsulates a cyclic hydrogen bonding network constructed by six “wing-tip” OH groups (Fig. 2c and d). A more fascinating feature of the dimeric (1U)2 is additional stabilization provided by six C–H⋯F contacts between the propargyl C–H groups and the fluorinated benzene core (Fig. 2a and c). The relatively short C⋯F distance of 3.228(8) Å observed here lies toward the shorter end of the values determined for previously reported structures.13a,13c–e,19–21
The functional relevance of such C–H⋯F contacts in the self-assembly process was evaluated further by density functional theory (DFT) computational studies on the simplified model compounds 1′ and 5 (at B3LYP/6-31G** theory level). The pseudo D3d-symmetric optimised geometry (Fig. S4, ESI‡) of (1′)2 provided an average value of d(F⋯H) = 2.45 Å along the C–H⋯F contacts, which is ca. 0.17 Å shorter than the sum of van der Waals radii of F and H.
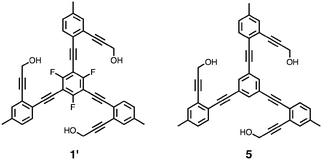
Energy evaluation at the B3LYP/cc-pVTZ(-f) level further supported the notion that such C–H⋯F interactions contribute to the thermodynamic stability of the dimer. Specifically, (1′)2 is stabilised by 7.4 kcal mol−1 relative to the essentially isostructural (5)2 lacking the C–F units (Fig. S5, ESI‡), when the energy difference between the individual monomers and the corresponding dimers was compared (Fig. S6, ESI‡). Divided by the total number of contacts within the dimer, each C–H⋯F interaction contributes to the overall stability by ca. 1.2 kcal mol−1.
Similarly to the self-association of 1U, its conformational isomer 1F also forms a dimeric structure in the solid state, but the topology of (1F)2 is determined by a quite different set of non-covalent interactions. As shown in Fig. 1b, 1F adopts a planar conformation, which is anticipated to promote cofacial stacking. Indeed, the assembly of dimeric (1F)2 is assisted by π–π stacking (ca. 3.38 Å) between electron-deficient fluorinated benzene core and electron-rich tert-butyl-substituted phenyl ring, the complementarity of which is further strengthened by the existence of two such contacts within the dimer (Fig. 3a–c).
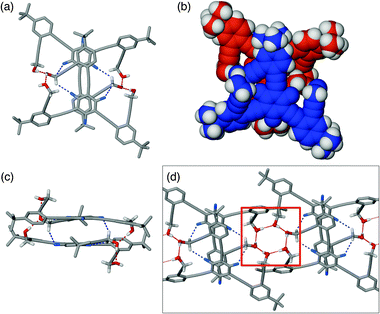 | ||
| Fig. 3 Capped stick and space-filling representations of the solid-state structure of dimeric (1F)2 generated using X-ray coordinates: (a) and (b) top view; (c) side view. In (d) is shown the extended structure of hydrogen-bonded (1F)2 with hexameric OH cluster motif highlighted within a red box. The O–H⋯O and C–H⋯F interactions are represented by dotted lines. | ||
In addition, the “dangling” OH group in the monomeric 1F (Fig. 1b) engages in an intermolecular hydrogen bond (O⋯O = 2.654(4); 2.670(4) Å) to the O–H⋯O–H unit of the stacking partner (Fig. 3a and c) to complete an extended O–H⋯O–H⋯O–H array. Intriguingly, this crescent-shaped O–H⋯O–H⋯O–H motif on each side of (1F)2 can further associate with the complementary O–H⋯O–H⋯O–H unit of the neighboring dimer to close up the six-membered cyclic hydrogen bond (Fig. 3d). The crystal structure of 1F also revealed an intermolecular C–H⋯F contact (C⋯F = 3.207(5) Å).
In summary, an intimate interplay of O–H⋯O/C–H⋯F hydrogen bonds and π–π stacking collectively gives rise to conformational pseudopolymorphs of a dendritic π-conjugated molecule (Scheme 1). For the highly symmetric and non-planar conformer 1U, a maximum number of O–H⋯O and C–H⋯F contacts is achieved by shape complementarity in monomer–monomer docking. For the less symmetric and largely planar conformer 1F, the molecule takes a different strategy to maximise monomer–monomer contacts by using predominantly a combination of π–π stacking and O–H⋯O interactions. Efforts are currently underway in order to prepare chiral derivatives of the parent system and to explore further the functional role of C–H⋯F hydrogen bonds in supramolecular chemistry.
This work was supported by the National Science Foundation (CAREER CHE 0547251) and DTRA/US Army Research Office (W911NF-07-1-053).
Notes and references
- (a) J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed; (b) R. J. Davey, Chem. Commun., 2003, 1463–1467 RSC; (c) G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007–1026 CrossRef CAS; (d) J. Bernstein, J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2008, 8, 2011–2018 CrossRef CAS; (e) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS; (f) L. Yu, Acc. Chem. Res., 2010, 43, 1257–1266 CrossRef CAS.
- A. Nangia, Cryst. Growth Des., 2006, 6, 2–4 CrossRef CAS and references cited therein.
- Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) H. G. Brittain, J. Pharm. Sci., 2009, 98, 1617–1642 CrossRef CAS; (b) A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS.
- (a) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, NY, 1997 Search PubMed; (b) L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2492 CrossRef CAS; (c) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- (a) G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS; (b) G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS; (c) P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS.
- (a) X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128–15139 CrossRef CAS; (b) C. Li, S.-F. Ren, J.-L. Hou, H.-P. Yi, S.-Z. Zhu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725–5729 CrossRef CAS; (c) C. Li, Y.-Y. Zhu, H.-P. Yi, C.-Z. Li, X.-K. Jiang, Z.-T. Li and Y.-H. Yu, Chem.–Eur. J., 2007, 13, 9990–9998 CrossRef CAS.
- S. A. Harrell and D. H. McDaniel, J. Am. Chem. Soc., 1964, 86, 4497–4497 CrossRef CAS.
- Understanding the supramolecular chemistry of C–F group is thus considered as one of the last frontiers in intermolecular interactions1e.
- J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS.
- J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89–98 CrossRef CAS.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS; (b) F. Hof and F. Diederich, Chem. Commun., 2004, 477–480 RSC.
- (a) V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS; (b) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS; (c) A. R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47–52 CrossRef; (d) A. Schwarzer, W. Seichter, H. Stoeckli-Evans, M. Losada and J. Hulliger, CrystEngComm, 2004, 6, 567–572 RSC; (e) M. Hyacinth, M. Chruszcz, K. S. Lee, M. Sabat, G. Gao and L. Pu, Angew. Chem., Int. Ed., 2006, 45, 5358–5360 CrossRef CAS.
- (a) X. Jiang, Y.-K. Lim, B. J. Zhang, E. A. Opsitnick, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2008, 130, 16812–16822 CrossRef CAS; (b) E. Opsitnick and D. Lee, Chem.–Eur. J., 2007, 13, 7040–7049 CrossRef CAS; (c) H. Y. Lee, X. Song, H. Park, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2010, 132, 12133–12144 CrossRef CAS.
- R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS.
- In analogy to protein secondary structures, the term “folding” here describes conformational restrictions imposed by non-covalent bonds.
- The structure of 1F is disordered over two positions, for which only one model is shown in Fig. 1 and 2. See ESI‡.
- The dihedral angle here is the angular relationship between the central C6F3 fragment and the peripheral aryl rings, all treated as least squares planes calculated from crystallographic data. In this analysis, τ = 90° corresponds to a perfectly orthogonal arrangement, whereas τ = 0° corresponds to a perfectly coplanar arrangement.
- A. R. Choudhury, K. Nagarajan and T. N. G. Row, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o644–o647 CrossRef CAS.
- A. J. Mountford, D. L. Hughes and S. J. Lancaster, Chem. Commun., 2003, 2148–2149 RSC.
- H.-C. Weiss, R. Boese, H. L. Smith and M. M. Haley, Chem. Commun., 1997, 2403–2404 RSC.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Details of the synthetic procedures, crystallographic data, and DFT calculations. CCDC 783201 and 783202. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c0cc02177f |
§ Crystal data. 1U (CCDC 783202) C53.27H47.27Cl6.82F3O3M = 1034.63, trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , a = 17.1496(12) Å, c = 33.798(2) Å, V = 8608.5(9) Å3, T = 150(2) K, Mo-Kα, 26 , a = 17.1496(12) Å, c = 33.798(2) Å, V = 8608.5(9) Å3, T = 150(2) K, Mo-Kα, 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 759 reflections, 3403 unique (Rint = 0.025). R1 = 0.0905, wR2 = 0.2683 (for 2596 observed reflections with I > 2σ(I) and 276 parameters, 784 restraints). 1F (CCDC 783201) C51H45F3O3M = 762.87, triclinic P 759 reflections, 3403 unique (Rint = 0.025). R1 = 0.0905, wR2 = 0.2683 (for 2596 observed reflections with I > 2σ(I) and 276 parameters, 784 restraints). 1F (CCDC 783201) C51H45F3O3M = 762.87, triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.4830(8) Å, b = 14.9544(12) Å, c = 15.7398(13) Å, α = 89.095(2)°, β = 86.983(2)°, γ = 71.805(2)°, V = 2340.9(3) Å3, T = 150(2) K, Mo-Kα, 29791 reflections, 7914 unique (Rint = 0.045). R1 = 0.0691, wR2 = 0.2059 (for 5124 observed reflections with I > 2σ(I) and 523 parameters, 12 restraints). The contribution of the diffuse contributing solvent (=two CHCl3 per molecule of 1F) to the structure factors was assessed by back-Fourier transformation, and the data were corrected accordingly using Platon/SQUEEZE. The sum formula of 1F excludes these solvent molecules. , a = 10.4830(8) Å, b = 14.9544(12) Å, c = 15.7398(13) Å, α = 89.095(2)°, β = 86.983(2)°, γ = 71.805(2)°, V = 2340.9(3) Å3, T = 150(2) K, Mo-Kα, 29791 reflections, 7914 unique (Rint = 0.045). R1 = 0.0691, wR2 = 0.2059 (for 5124 observed reflections with I > 2σ(I) and 523 parameters, 12 restraints). The contribution of the diffuse contributing solvent (=two CHCl3 per molecule of 1F) to the structure factors was assessed by back-Fourier transformation, and the data were corrected accordingly using Platon/SQUEEZE. The sum formula of 1F excludes these solvent molecules. |
| This journal is © The Royal Society of Chemistry 2011 |