Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels
Juan Carlos
Serrano-Ruiz
*a and
James A.
Dumesic
b
aAdvanced Materials Laboratory, Department of Inorganic Chemistry, University of Alicante, Apartado 99, 03080, Alicante, Spain. E-mail: jcserrano@ua.es; Fax: +34 965 903 454; Tel: +34 965 903 400 ext. 2048
bDepartment of Chemical and Biological Engineering, University of Wisconsin–Madison, WI 53706, USA. E-mail: dumesic@engr.wisc.edu; Fax: +1 608 262 5434; Tel: +1 608 262 1095
First published on 30th November 2010
Abstract
Concerns about diminishing fossil fuel reserves along with global warming effects caused by increasing levels of CO2 in the atmosphere are driving society toward the search for new renewable sources of energy that can substitute for coal, natural gas and petroleum in the current energy system. Lignocellulosic biomass is abundant, and it has the potential to significantly displace petroleum in the production of fuels for the transportation sector. Ethanol, the main biomass-derived fuel used today, has benefited from production by a well-established technology and by partial compatibility with the current transportation infrastructure, leading to the domination of the world biofuel market. However, ethanol suffers from important limitations as a fuel (e.g., low energy density, high solubility in water) than can be overcome by designing strategies to convert non-edible lignocellulosic biomass into liquid hydrocarbon fuels (LHF) chemically similar to those currently used in internal combustion engines. The present review describes the main routes available to carry out such deep chemical transformation (e.g., gasification, pyrolysis, and aqueous-phase catalytic processing), with particular emphasis on those pathways involving aqueous-phase catalytic reactions. These latter catalytic routes achieve the required transformations in biomass-derived molecules with controlled chemistry and high yields, but require pretreatment/hydrolysis steps to overcome the recalcitrance of lignocellulose. To be economically viable, these aqueous-phase routes should be carried out with a small number of reactors and with minimum utilization of external fossil fuel-based hydrogen sources, as illustrated in the examples presented here.
 Juan Carlos Serrano-Ruiz | Dr Juan Carlos Serrano-Ruiz studied Chemistry at the University of Granada (Spain). In 2001 he moved to the University of Alicante (Spain) where he received a PhD in Chemistry and Materials Science in 2006 under the supervision of Prof. Francisco Rodríguez-Reinoso and Antonio Sepúlveda-Escribano. In January 2008 he was awarded a Fulbright fellowship to conduct studies on biomass conversion to fuels and chemicals by catalytic approaches in Prof. James Dumesic's research group at the University of Wisconsin–Madison, Madison (USA). In January 2010 he joined the Advanced Materials Laboratory (LMA) at the University of Alicante where he has initiated a new research line on catalytic routes for the conversion of biomass into liquid hydrocarbon fuels. He is member of the Spanish Biomass Technology Platform (BioPlat) and he belongs to the Spanish Biofuels for Transport Working Group. |
 James A. Dumesic | James A. Dumesic earned his BS degree from UW–Madison and his MS and PhD degrees from Stanford University, under the supervision of Professor Michel Boudart. Dumesic joined the Department of Chemical Engineering in 1976, and he is currently the Steenbock Chair in the College of Engineering. He has recently studied how aqueous-phase reforming of glucose or sorbitol can be tailored to selectively produce H2 or directed to produce hexane. He has investigated methods to produce liquid alkanes by acid catalyzed dehydration of sugars, followed by aldol-condensation over solid base catalysts. He has shown how that liquid alkanes can be produced from glycerol via an integrated process involving catalytic conversion to H2/CO gas mixtures and Fischer–Tropsch synthesis in a single reactor, and he has studied strategies for catalytic conversion of sugars and polyols to hydrocarbons by first producing monofunctional intermediates over PtRe/C catalysts, followed by catalytic upgrading to control the extent of C–C coupling. Most recently, he has been studying the use of levulinic acid and γ-valerolactone as biomass-derived platform chemicals for the production of fuels and chemicals. |
Broader contextThe production of liquid transportation fuels from renewable biomass sources is a promising route that can help to decrease our dependence on petroleum and to mitigate environmental issues. The transportation sector of our society and its entire infrastructure is based on petroleum-derived liquid hydrocarbons, the latter of which possess high energy density and optimum combustion characteristics. First-generation biofuels (i.e., ethanol and biodiesel) are produced from food crops, and present different chemical compositions than conventional hydrocarbon fuels, creating important compatibility and energy density issues. One promising alternative to overcome these limitations of oxygenated fuels is to use non-edible biomass (i.e., lignocellulose) to produce liquid hydrocarbon fuels chemically similar to those currently used in cars, trucks or aircraft. The present paper describes the most promising routes to achieve this transformation, with special emphasis on those approaches involving catalytic reactions. |
1. Introduction
Society has reached high levels of development during the last century. This progress, however, has been achieved at the expense of extensive consumption of natural resources, such as petroleum, natural gas and coal. These fossil fuel resources took millions of years to be formed, and they are currently being consumed at a rate that is orders of magnitude higher than their natural regeneration cycle, making them non-renewable sources of energy. The most recent data available for world energy consumption indicate that society still remains highly dependent on fossil fuels at the present time. For example in 2008, fossil fuels supplied 85% of the total energy consumed in the US,1 and almost 80% of the energy produced in the European Union.2 These fossil fuel resources are used to provide energy for various sectors of society (i.e., residential, commercial, industrial, transportation and electrical power), among which the transportation sector is the largest and fastest growing energy sector, responsible for almost one third of the total energy consumed in the world. Moreover, a large fraction of the energy for the transportation sector (96%) is currently derived from petroleum.3Three important issues are associated with the large-scale utilization of fossil fuels: availability, global warming and uneven geographic distribution of reserves. Fossil fuels are finite and, as indicated above, their current consumption rate is higher than their corresponding regeneration rate, leading inevitably to depletion. Projections for the near future indicate that world energy consumption will increase by 35% over the next 20 years to meet the growing demand of industrialized countries and the rapid development of emerging economies,4 and world demand for petroleum will raise by 30%, reaching 111 millions of barrels per day in 2035.5 Taking into account these forecasts and current data of proven reserves, it has been estimated that oil, natural gas and coal will be depleted within the next 40, 60 and 120 years, respectively.6 In the case of petroleum, many researchers predict a more dramatic situation and estimate that the global production of oil will reach a maximum in the year 2020 and decay thereafter.7
Global warming is, possibly, the most dramatic and known collateral effect produced by the massive utilization of fossil fuels.8 Fossil fuels are transformed into energy by means of combustion reactions, leading to net emissions of CO2, a strong greenhouse gas, into the atmosphere. Accordingly, the extraction of fossil fuels for energy production has allowed a large part of the carbon stored in the earth for millions of years to be released in just a few decades.
Fossil fuels reserves are not equally distributed around the world. The Middle-East countries control the 60% of the oil reserves and the 41% of natural gas supplies, and only three countries (US, China and Russia) account for 60% of the world recoverable coal reserves.4 This situation can lead to economic instabilities, requires the transportation of fossil fuel resources over long distances, and can cause political and security problems worldwide.
The issues outlined above, inherently associated with fossil fuels, suggest that society requires new sources of energy to ensure progress and protect the environment for future generations. These new sources of energy should: (i) have the potential to effectively replace fossil fuels in the current energy production system and (ii) be renewable, well distributed around the world, and not contribute to the accumulation of greenhouse gases into the atmosphere. In this respect, natural resources such as solar energy, wind, hydroelectric power, geothermal activity, and biomass meet these requirements. Unlike fossil fuels, they are abundant and allow the development of zero-carbon or carbon-neutral technologies, thus contributing to mitigation of global warming effects. Substitution of fossil fuel-based technologies for those derived from renewable sources is currently spurred by various governments,9,10 and it will be done progressively and selectively. Thus, while solar, wind, hydroelectric, and geothermal have been proposed as excellent alternatives to coal and natural gas for heat and electricity production in stationary power applications,11,12 biomass is the only sustainable source of organic carbon currently available on earth,13 and it is considered to be an ideal substitute for petroleum in the production of fuels, chemicals and carbon-based materials.14,15 However, when designing strategies for potential replacement of crude oil by biomass, it is important to note that the petrochemical industry currently consumes three quarters of the crude oil to cover the demand for liquid hydrocarbon fuels of the transportation sector, whereas only a small fraction of the petroleum is utilized in the synthesis of industrial chemicals and other derivatives.16 Consequently, an effective implementation of biomass in the current energy system will necessarily involve the development of new technologies for the large-scale production of biofuels.
At the present time, two biomass-derived fuels (so-called first generation of biofuels) have been successfully implemented in the transportation sector: biodiesel (a mixture of long-chain alkyl esters produced by transesterification of vegetable oils with methanol) and ethanol (produced by bacterial fermentation of corn and sugar cane-derived sugars). The penetration of these liquid biofuels in the transportation sector is still very weak, and in 2005 they represented only 2% of the total transportation energy.3 However, the important environmental and economic benefits derived from their large-scale utilization will stimulate society to progressively increase reliance on biofuels. Thus, according to projections by the International Energy Agency, the world biofuel production will increase from the current level of 1.9 million of barrels per day (mbd) in 2010 to 5.9 mbd by 2030, which represents 6.3% of the world conventional fuels production.4 Unlike petroleum-based fuels, liquid biofuels are considered carbon neutral since CO2 produced during fuel combustion is consumed by subsequent biomass regrowth.17 Furthermore, recent studies indicate that the use of liquid biofuels produced domestically would strengthen economies by reducing the dependence of foreign oil and by creating new well-paid jobs in different sectors such as agricultural, forest management and oil industries.18
2. Lignocellulosic liquid hydrocarbon fuels: alternative to ethanol
The current biofuel market is largely dominated by ethanol, which accounts for 90% of world biofuel production.19 Indeed, the rate of ethanol production around the world is increasing rapidly, from 13 billion gallons in 2007 to the current level of almost 20 billion gallons in 2009,20 with the US (55%, corn-derived) and Brazil (33%, sugar cane-derived) being the main producers.21 The ethanol industry has benefited from a mature and simple technology in which selected microorganisms (e.g., yeast, bacteria, and mold) transform aqueous sugars to the desired final ethanol product. Ethanol is added to gasoline to increase the octane number of the mixture and, with it, improve the combustion characteristics of the fuel. The oxygen in ethanol allows low-temperature combustion with the subsequent reduction of pollutants such as CO and NOx.19 Additionally, apart from CO2 emission savings, blending gasoline with ethanol helps to reduce SOx emissions to the atmosphere, because ethanol contains a negligible amount of sulfur compared to petroleum.18A key aspect that is responsible for expansion of the ethanol industry in recent years is the compatibility of ethanol with the existing infrastructure for gasoline. Thus, ethanol blended with conventional gasoline is currently used in many countries as a renewable fuel in existing spark-ignition engines. This compatibility, however, is not complete and the use of ethanol is presently limited to low-concentration blends (5–10% by volume), namely E5–E10. Ethanol-enriched mixtures such as E85 require cars with specially designed engines, designated as flexible-fuel vehicles (FFVs), which are commonly used only in a few countries like Brazil and Sweden. E85 mixtures are not tolerated by conventional vehicles, because ethanol, especially in high-concentration blends, can cause corrosion of some metallic components in tanks and deterioration of rubbers and plastics used in internal combustion engines.21 This constraint in ethanol blending represents the main issue of the growing ethanol industry. As outlined in Fig. 1, projections indicate that the US ethanol industry will approach the blending wall (i.e., the point at which blending 10% of ethanol in each gallon of gasoline will not be able to accommodate the rate of ethanol production) in 2010.22 Furthermore, experts predict that the number of E85 fuelling stations and flexi-fuel vehicles will not grow sufficiently fast to accommodate the growing volumes of ethanol produced in the US.23 A potential solution to overcome the blending wall is to raise the amount the ethanol allowed in gasoline to beyond 10% by commercializing intermediate ethanol blends (i.e., E15–E20) (Fig. 1). However, there are serious issues in using blends with higher ethanol concentrations. In countries like the US, the utilization of E15–E20 blends in regular vehicles is still not authorized, since the effect of these mixtures on pollutant emissions, driving performance and materials compatibility (e.g., tanks, pipelines, dispensers) is not fully understood,23 and current European standards allow only for E5 blends.18
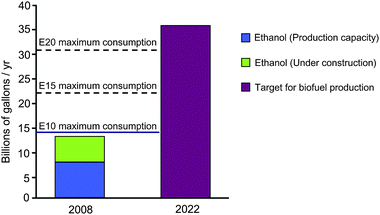 | ||
| Fig. 1 Volumes of ethanol absorbed by several blends in the US. Target for biofuels production refers to the level of biofuels production mandated by the Energy Independence Security Act of 2007 (EISA 2007).26 Maximum consumption for a determined blend refers to the ethanol consumed if all the gasoline used in the country is blended with ethanol in the amount indicated. Source: Biomass Research and Development Board.23 | ||
Apart from the aforementioned blending issues, ethanol presents another important limitation as a transportation fuel. Ethanol contains less energy per volume (i.e., energy density) than conventional gasoline, which ultimately reduces the fuel mileage of the vehicles. In this sense, it has been estimated that cars running on ethanol rich mixtures like E85 operate with 30% lower fuel mileage.24 This fact, along with the small price differential between E85 and regular gasoline, has discouraged drivers to purchase E85 cars or fuel so far.22
Conventional transportation fuels are composed of liquid hydrocarbons with different molecular weights (e.g., C5–C12 for gasoline, C9–C16 for jet fuel, and C10–C20 for diesel applications) and chemical structures (e.g., branched for gasoline, linear for diesel). The entire transportation infrastructure (including engines, fueling stations, distribution networks, and storage tanks) has been developed to take advantage of the excellent properties of these compounds as fuels. Thus, the special composition of hydrocarbons fuels, based only on carbon and hydrogen, provides them with high energy-density and stability (allowing efficient storage at ambient conditions) and superior combustion characteristics, properties highly desired for transportation liquids. Thus, instead of using biomass to produce oxygenated fuels (such as ethanol) with new compositions, an attractive alternative would be to utilize biomass to generate liquid fuels chemically similar to those being used today derived from oil.17,25 These new fuels would be denoted as green gasoline, green diesel and green jet fuel, and they would be essentially the same as those currently used in the transportation fleet, except that they would be synthesized from biomass instead of petroleum. When compared with ethanol, the production of hydrocarbon fuels from biomass has important advantages. The main benefit would include full compatibility with the existing energy system. Since green hydrocarbon fuels would be essentially the same as those currently obtained from petroleum, it would not be necessary to modify engines, pumps or distribution networks to accommodate the new renewable liquids in the transportation sector.
Unlike ethanol, biomass-based hydrocarbons fuels are energy equivalent to fuels derived from petroleum. The heating value (i.e., the heat released when a known quantity of fuel is burned under specific conditions) of ethanol is only two-thirds that of gasoline, which, as indicated above, penalizes the fuel mileage of the vehicles running on gasoline–ethanol mixtures. The use of renewable hydrocarbon fuels would additionally help to meet the increased standards of fuel economy imposed by governments to the automobile industry. In the case of the US, these standards establish a mandatory increase in average fuel economy from the current 25 miles per gallon (mpg) to 35 mpg by 2022.26
The addition of oxygenated components to conventional fuels increases the water solubility of the mixture. This increase is particularly marked in the case of gasoline–ethanol blends, since pure ethanol is highly hygroscopic and completely miscible in water. Thus, adding 10% of ethanol to regular gasoline raises the water solubility of the blend more than 30 times (from 150 ppm v/v of regular gasoline to 5000 ppm for E10).27 Once the water contamination reaches the saturation level, additional water separates from the mixture, removing the ethanol from gasoline and leading to phase separation. In fact, when phase separation occurs in the storage tank, the ethanol–water layer may combust in the engine at higher temperatures causing damage to it.27 The water tolerance of a gasoline–ethanol blend (i.e., fraction of water that the mixture can contain without phase separation) decreases with temperature and increases with ethanol content (Fig. 2). Consequently, phase separation is an important concern in countries with cooler climates and when low-concentration blends such as E5 are used. Water can be absorbed by the ethanol–gasoline mixture from the atmosphere (in the form of moisture), from the air trapped in the tank (by condensation of water when temperature decreases), or even from the ethanol itself which typically carries traces of water when delivered from the biorefinery. In this respect, many countries have regulated the maximum amount of water allowed in fuel-grade ethanol to the level of 1% (v/v), to avoid phase separation issues.28 Ethanol affinity for water has important implications for distribution logistics as well. Pipelines, considered to be the least expensive means of safely transporting bulk fuel shipments,23 are not suited to transport ethanol or gasoline–ethanol blends on a commercial scale, because apart from corrosion issues, ethanol can pick up water in the pipeline with the potential result of phase-separation. Consequently, ethanol has to be distributed by other fossil fuel-consuming transportation modes such as rail, truck and barge. The hydrophobic character of biomass-derived hydrocarbons eliminates these problems, since these molecules are immiscible in water. Additionally, the ability of liquid hydrocarbons to self-separate from water, as represented in Fig. 3, is highly beneficial in that it eliminates the need for expensive and energy-consuming distillation steps required in the ethanol purification process. In particular, ethanol is initially obtained in form of a dilute aqueous solution (5–12% v/v), which is subsequently concentrated to 96–99% by distillation. It is estimated that this intense water removal step is responsible for 35–40% of the total energy required for ethanol production,29 and this energy is typically supplied by combustion of fossil fuels such as natural gas.
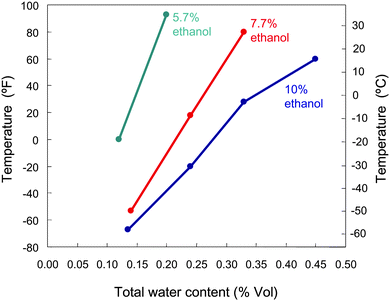 | ||
| Fig. 2 Water tolerance of some gasoline–ethanol blends as a function of temperature. Adapted from ref. 27. | ||
 | ||
| Fig. 3 Picture showing spontaneous separation between aqueous phase and liquid alkanes produced by processing of biomass-derived molecules in a four-phase flow reactor. Source: Dumesic's Research Group web site, http://jamesadumesic.che.wisc.edu/index2.htm. | ||
Any technology envisaged to convert biomass feedstocks into liquid fuels must address one important limitation of this resource: the low energy-density of biomass compared to fossil fuels. Although the energy-density of biomass varies considerably depending on the source, an average value for biomass (15–20 MJ kg−1) is well below that of crude oil (42 MJ kg−1).30 Large amounts of biomass will thus be required to produce liquid fuels, leading to high costs for transporting the biomass source to the processing location.31 Furthermore, if biomass transportation involves utilization of fossil fuels, then the overall CO2 emission savings of the bioprocess would be penalized. Consequently, for biomass conversion technologies to be cost-competitive and truly carbon-neutral, it is necessary to develop efficient processing units at small scale that can be distributed close to the biomass source.17 Even though ethanol plants achieve a significant size reduction compared to petrochemical refineries, the mild conditions employed and the low levels of ethanol achieved for bacterial fermentation (e.g., 30–50 °C, ethanol concentrations lower than 15% v/v) significantly limit the reaction rates and require reactors with a size large enough to make the process economically feasible. As will be described in following sections, biomass-based hydrocarbon fuels, in contrast, can be produced at high temperatures and using concentrated water solutions,14 which allow for faster conversions in smaller reactors.
The utilization of edible biomass (such as corn or cane sugar) for the large-scale production of fuels can produce competition with food for land use. The so-called food-versus-fuel debate has arisen in many countries as a response to the sharp increase in food prices during 2007 and 2008. Although it has been pointed out that this rise in price was the result of several linked worldwide events,22 some authors indicate that the increased demand for corn to produce ethanol had a direct impact on food prices, especially in food-insecure areas of the world where food is based on grain consumption.32 These issues have driven researchers around the world to develop technologies to process non-edible biomass (e.g., lignocellulosic biomass), thereby permitting sustainable production of a new generation of biofuels (so-called second generation of fuels), without affecting food supplies. Lignocellulosic biomass has two important advantages over edible biomass feedstocks: it is more abundant and can be grown faster and with lower costs.33 In this respect, it is estimated that the US could sustainably produce more than 1 billion tons of non-edible biomass per year by 2050 with relatively modest changes in land use and agricultural and forestry practices. Once converted into biofuels, these lignocellulosic resources would have the potential to displace more than one third of the petroleum currently consumed by the transportation sector.34 Lignocellulosic feedstocks ($3 per GJ) are slightly less expensive than edible biomass (5$ per GJ), and potentially more economical than crude oil (10–15 $ per GJ) and vegetable oils (18–20 $ per GJ); however, due to its recalcitrant nature, lignocellulose is more difficult to convert and, consequently, processing costs increase for this resource.35 Thus, according to recent analyses, the cost of producing lignocellulosic ethanol would be almost double that of corn-derived ethanol.19 This fact represents the main limitation of lignocellulose as a renewable resource, and the lack of cost-competitive technologies for the generation of liquid fuels from non-edible sources has been identified by experts as the key bottleneck for the large-scale implementation of lignocellulose-derived biofuel industry.36
Recalcitrance of lignocellulosic biomass can be explained in terms of its chemical structure, comprised of three major units: cellulose, hemicellulose and lignin.13,37,38 Cellulose (40–50%) is a high molecular weight polymer of glucose units connected linearly via β-1,4-glycoside linkages. This arrangement allows for extensive hydrogen bonding between cellulose chains, which confers this material with rigid crystallinity and, thus, high resistance to deconstruction.39 Cellulose bundles are additionally attached together by hemicellulose (15–20%), an amorphous (and consequently more readily deconstructed) polymer of five different C5 and C6 sugars. Cellulose and hemicellulose, the carbohydrate fraction of lignocellulose, are protected by a surrounding three-dimensional polymer of propyl-phenol called lignin (15–25%), which provides extra rigidity to the lignocellulose structure. To overcome lignocellulose recalcitrance, a variety of physical and chemical methods have been developed, and a comprehensive description of such technologies can be found elsewhere.40–42 The approach most commonly used involves pretreatment of lignocellulose (with the aim of breaking/weakening the lignin protection and increasing the susceptibility of crystalline cellulose to degradation), followed by hydrolysis to depolymerize hemicellulose and cellulose and, thus, isolate the sugars from the lignin fraction.
3. Routes for the production of biofuels from biomass
The main pathways for the production of liquid transportation fuels from biomass are shown in Fig. 4. As indicated in previous sections, food crops such as corn grain or sugar cane can be converted into ethanol by fermentation processes. Alternatively, second generation ethanol can be produced from lignocellulosic sources by means of pretreatment–hydrolysis and subsequent fermentation of soluble sugars. Butanol, with energy density and polarity similar to gasoline, can be also produced by this route, representing an interesting alternative to overcome many of the technical shortcomings of ethanol as a fuel.43,44 Interestingly, the microorganisms utilized for fermentation can be engineered to convert sugars to liquid alkanes instead of alcohols.45 This new technology could achieve improvements over classical fermentation approaches, because hydrocarbons separate spontaneously from the aqueous phase, thereby avoiding poisoning of microbes by the accumulated products and facilitating separation/collection of alkanes from the reaction medium.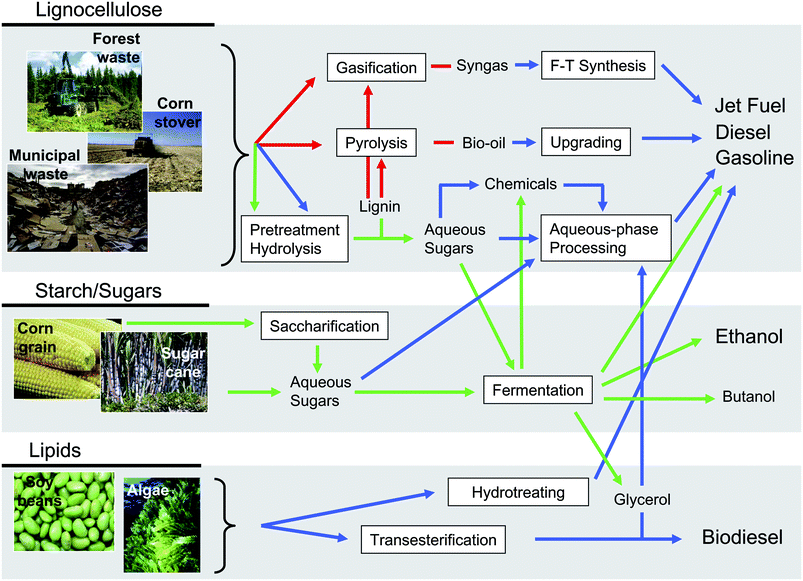 | ||
| Fig. 4 Routes for the conversion of biomass into liquid fuels. Red arrows refer to thermal routes, green arrows refer to biological routes, and blue arrows refer to catalytic routes. Adapted from ref. 25. | ||
Vegetable oils, obtained from food sources such as soybeans, palm or sunflower, can serve as feedstocks for the production of first-generation biodiesel through transesterification processes. Since vegetable oils are expensive and compete with food sources, the challenge of the biodiesel industry is to find non-edible sources of lipids. Algae crops are receiving interest in this respect,46 although the high cost associated with feedstock production is an important barrier, and related technologies are presently at an early stage of development. Green diesel can be produced from plant oils and animal fats by means of deoxygenation reactions under hydrogen pressure in hydrotreating processes.47,48 This recent technology has potential in that it can be carried out in existing petroleum refinery infrastructure.49
Representative examples of non-food lignocellulosic feedstocks such as forest wastes, agricultural residues like corn stover, or municipal paper wastes are shown in Fig. 4. Apart from their intrinsic recalcitrance, these feedstocks are characterized by a high degree of chemical and structural complexity, and, consequently, technologies for the conversion of these resources into liquid hydrocarbon fuels typically involve a combination of different processes. The methodology most commonly used to overcome lignocellulose complexity involves the transformation of non-edible feedstocks into simpler fractions that are subsequently more easily converted into a variety of useful products. This approach, similar to that used in conventional petroleum refineries, would allow the simultaneous production of fuels, power, and chemicals from lignocellulose in an integrated facility denoted as a biorefinery.50,51 Current technologies for converting lignocellulose to liquid hydrocarbon transportation fuels involve three major routes: gasification, pyrolysis and pretreatment–hydrolysis (Fig. 4). By means of these primary routes, lignocellulose is converted into gaseous and liquid fractions that are subsequently upgraded to liquid hydrocarbon fuels. Thus, gasification converts solid biomass to synthesis gas (syngas), a valuable mixture of CO and H2 which serves as a precursor of liquid hydrocarbon fuels by Fischer–Tropsch (F–T) reactions. This pathway is commonly known as biomass to liquids (BTL). Pyrolysis allows transformation of lignocellulosic biomass into a liquid fraction known as bio-oil that can be subsequently upgraded to hydrocarbon fuels by a variety of catalytic processes. The third route involves pretreatment–hydrolysis steps to yield aqueous solutions of C5 and C6 sugars derived from lignocellulose. While gasification and pyrolysis are pure thermal routes in which lignocellulose is decomposed with temperature under controlled atmosphere, aqueous-phase processing, in contrast, involves a series of catalytic reactions to selectively convert sugars and important platform chemicals derived from them into targeted liquid hydrocarbon fuels with molecular weights and structures appropriate for gasoline, diesel and jet fuel applications.
4. Production of liquid hydrocarbon transportation fuels from lignocellulose
4.1 Biomass to liquids (BTL)
Biomass to liquids can be described as a renewable version of fossil fuel-based technologies like coal to liquids (CTL) and gas to liquids (GTL), involving the integration of two different processes: biomass gasification to syngas (H2/CO) and F–T synthesis. Even though both technologies are well known and relatively mature, integration remains a challenge in BTL, because the utilization of lignocellulosic biomass as a feedstock (in substitution for classical carbon sources such as coal and natural gas) introduces new difficulties in the overall process. Biomass gasification is achieved by treatment at high temperatures (e.g., 1100–1500 K) under a well-controlled oxidizing atmosphere (e.g., air, steam, oxygen). Control over the composition of the outlet gaseous stream is difficult and depends on a variety of factors including the oxidizing agent, biomass particle size and gasifier design.52 In this respect, research indicates that utilization of pure oxygen atmosphere, small particle sizes (lower than 1 mm diameter), and a combination of high temperatures, high pressures and low residence times favors the production of syngas versus producer gas (a mixture of CO, H2, CO2, CH4, and N2 used for heat and electricity production).53–55The direct integration of biomass gasification and F–T synthesis requires an intermediate gas-cleaning system, because the gaseous stream delivered from the gasifier typically contains a number of contaminants that need to be removed before the F–T unit, which is highly sensitive to impurities. Thus, tars (condensable high molecular weight hydrocarbons produced by incomplete biomass gasification), volatile species such as NH3, HCl, and sulfur compounds (produced by gasification of lignocellulose impurity components), fine particles, and ashes typically accompany CO and H2 in the outlet gaseous stream. The high number of contaminants, along with the strict cleaning standards imposed by the F–T unit,56 require the use of multiple steps and advanced technologies52 that contribute significantly to the complexity and cost of the BTL plant. Additionally, because biomass contains higher amounts of oxygen compared to coal, the syngas delivered from lignocellulosic sources is typically enriched in CO (H2/CO = 0.5), and F–T synthesis requires syngas with a H2/CO ratio closer to 2.57,58 By providing sufficient water co-feeding, the H2/CO ratio can be adjusted by means of an intermediate water gas-shift (WGS, CO + H2O → CO2 + H2) reactor situated between the gasifier and the F–T unit.
The F–T reactor, the last unit of the BTL plant, achieves conversion of syngas to a distribution of alkanes over Co-, Fe-, or Ru-based catalysts in a well-developed industrial process.58 However, the hydrocarbons produced by the direct route range from C1 to C50, and neither gasoline nor diesel fuels can be produced selectively without generating a large amount of undesired products. Indirect approaches involve initial production of heavy hydrocarbons (waxes), followed by controlled cracking of the heavy compounds to diesel and gasoline components to overcome this limitation.59
The cost of producing the biofuel, negatively affected by the complexity of the process, is the main factor limiting the commercialization of BTL technologies. Application of economies-of-scale allows for improvements in the economics of the process at the expense of having large centralized facilities that, as indicated in a previous section, lead to higher costs for transporting the low energy density biomass. BTL profit margins can be increased by co-producing, along with liquid hydrocarbon fuels, higher-value chemicals such as methanol60 and hydrogen61,62 from lignocellulose-derived syngas. Another positive aspect of BTL is its versatility. Thus, since any source of lignocellulose can be potentially gasified, BTL technologies are not constrained to a particular biomass feedstock or fraction.
4.2 Pyrolysis integrated with upgrading processes
Lignocellulosic biomass can be treated under inert atmosphere at temperatures of 648–800 K in a process called pyrolysis. At these conditions, solid biomass undergoes a number of processes including depolymerization, dehydration and C–C bond breaking reactions which lead to the formation of reactive vapor species.35 Upon subsequent cooling, the vapor products condense generating a dark viscous liquid referred to as bio-oil. This bio-oil is a complex mixture of more than 400 highly oxygenated compounds, including acids, alcohols, aldehydes, esters, ketones and aromatic species, along with some remnants of polymeric carbohydrates and lignin fragments.63,64 Consequently, once separated into their components, bio-oil could serve as a source of chemicals. The final composition of the bio-oil depends on a large number of factors (e.g., feedstock type, reaction conditions, alkali content of the feedstock, storage conditions). Bio-oils typically contain about 25 wt% water (derived from the initial water of the feedstock and from the pyrolysis process itself), and retain up to 70% of the energy stored in the biomass feedstock,52 thereby allowing for concentration of the energy of biomass in a dense liquid that is more easily transportable. The main advantage of pyrolysis over BTL is its simplicity, because it requires only a single reactor and low capital investments, thereby allowing the development of cost-effective processing units on small scale. Thus, small portable pyrolysis reactors (i.e., 50–100 tons of biomass per day) are currently commercially developed to produce liquid biofuels close to the biomass location in countries like the US, Canada and the Netherlands.65,66Even though bio-oils can be used directly in simple boilers and turbines for heat and electricity production, their utilization as transportation fuels has multiple shortcomings. The high oxygen content of bio-oils negatively affects the energy density (16–19 MJ kg−1versus 46 MJ kg−1 of regular gasoline), and it leads to low volatility and poor stability properties of the bio-oil liquid. Furthermore, the high corrosiveness (pH ≈ 2.5) and viscosity of bio-oils discourage their utilization in internal combustion engines. Since the pyrolysis process does not involve a deep chemical transformation in the feedstock, extensive oxygen removal is required for bio-oils to have hydrocarbon-like properties (e.g., high energy density, high volatility and high thermal stability), and several routes are available in this respect (Fig. 5).
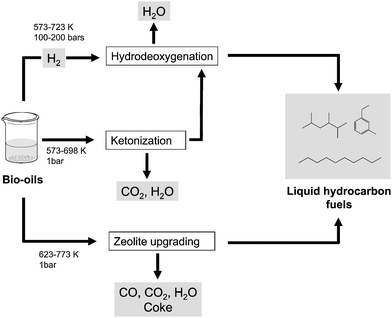 | ||
| Fig. 5 Catalytic routes for the upgrading of biomass-derived oils into liquid hydrocarbon transportation fuels. | ||
Hydrodeoxygenation (i.e., treatment of the bio-oil at moderate temperatures and high hydrogen pressures, HDO) is probably the most common method to achieve oxygen removal from bio-oils.67,68 By means of this technology, bio-oil components are completely hydrogenated and oxygen is removed in the form of water, which appears in the reactor as a separate phase from the hydrocarbon layer. Hydrodeoxygenation is typically carried out over sulfided CoMo and NiMo based catalysts68 (used in the petrochemical industry to achieve sulfur and nitrogen removal from crude oil). Precious metals such as Pt and Ru69,70 show higher hydrogenation activities at the expense of low tolerance to sulfur impurities (typically present in bio-oils). The large amount of hydrogen required for bio-oil deoxygenation represents the main drawback of this technology,71 and strategies based on steam-reforming of the water-soluble fraction of bio-oils,72 along with aqueous-phase reforming of biomass-derived sugars,73,74 have been studied to avoid the need to supply hydrogen from external fossil fuel sources. Bio-oils typically contain significant amounts of lignin-derived phenols which, once transformed into aromatic hydrocarbons, are valuable gasoline components.75 One of the challenges of the hydrodeoxygenation process is to achieve complete hydrogenation of aliphatic compounds while avoiding unnecessary hydrogen consumption in the reduction of the valuable aromatic hydrocarbons. However, this control over the extent of the hydrogenation process is difficult at the elevated hydrogen pressures required for hydrodeoxygenation (e.g., 100–200 bars). In addition, high pressures lead to increases in operational costs of the process.
Bio-oil deoxygenation can be alternatively carried out at milder conditions (e.g., 623–773 K, atmospheric pressure) and without external hydrogen by processing the bio-liquid over acidic zeolites, in a route that resembles the catalytic cracking approach used in petroleum refining.76–78 At these conditions, bio-oil components undergo a number of reactions involving dehydration, cracking and aromatization, and oxygen is removed in the form of CO, CO2 and water (Fig. 5). As a result, bio-oil is converted into a mixture of aliphatic and aromatic hydrocarbons, although a large fraction of the organic carbon reacts to form solid carbonaceous deposits denoted as coke. Thus, hydrocarbon yields are relatively modest and regeneration cycles under air (to burn off the coke) are frequent. Irreversible deactivation, caused by partial de-alumination of zeolite structures at the water contents typically found in bio-oils, is another drawback of this technology, and research is needed on new acidic catalytic materials with better resistance to water.16 On the other hand, the conditions of pressure and temperature at which zeolite upgrading is carried out are similar to those used in pyrolysis, thereby allowing the integration of these two processes in a single reactor, as recently demonstrated by Huber et al.79
A third route that could help to reduce oxygen content in bio-oils while leaving the bio-liquid more amenable for subsequent downstream processes is catalytic ketonic decarboxylation or ketonization80 (Fig. 5). By means of this reaction, 2 molecules of carboxylic acids are condensed into a larger ketone (2n − 1 carbon atoms) with the release of stoichiometric amounts of CO2 and water. This reaction is typically catalyzed by inorganic oxides such as CeO2, TiO2, Al2O3 and ZrO2 at moderate temperatures (573–698 K) and atmospheric pressure.81–84 Interestingly, ketonization achieves oxygen removal (in the form of water and CO2) while consuming carboxylic acids, the latter of which represent an important fraction of bio-oils (up to 30 wt%).85 Moreover, these acids are hydrogen-consuming compounds, and are responsible for unwanted properties of the bio-liquids such as corrosiveness and chemical instability. Consequently, as represented in Fig. 5, a pretreatment of the bio-oil over a ketonization bed would simultaneously reduce oxygen content and acidity, thereby reducing hydrogen consumption and leaving bio-oil more amenable for subsequent hydrodeoxygenation processing. Even though ketonization has not been used to process real lignocellulosic bio-oils so far, we believe that this route has potential to upgrade bio-liquids enriched in carboxylic acids. Furthermore, ketonization can also condense typical components of bio-oils like esters,86–88 and, unlike zeolite upgrading, this reaction can be efficiently carried out under moderate amounts of water.89
4.3 Aqueous-phase processing of biomass derivatives
As indicated in Fig. 4, aqueous solutions of sugars, derived from the carbohydrate fraction of lignocellulose (i.e., cellulose and hemicellulose), can be used to produce second generation ethanol fuel through fermentation routes. Alternatively, these sugars can be transformed into a variety of useful derivatives by means of chemical and biological processes.90,91 As will be addressed in this section, sugars and some of their derivatives can be catalytically processed in the aqueous phase to produce liquid fuels chemically identical to those currently used in the transportation sector. The key advantage of this route, in comparison with BTL and pyrolysis-upgrading approaches, is derived from the mild reaction conditions used, allowing for better control of conversion selectivity. However, costly pretreatment and hydrolysis steps are required to hydrolyze solid lignocellulose to soluble sugar feeds, and the lignin fraction, once isolated, is typically combusted to provide heat and power.The production of liquid hydrocarbon transportation fuels from biomass derivatives involves deep chemical transformations. In this respect, sugars (and chemicals derived from them) are molecules with high degrees of functionality (e.g., –OH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH groups) and a maximum number of carbon atoms limited to six (derived from glucose monomers). On the other hand, hydrocarbon fuels are larger (up to C20 for diesel applications) and completely unfunctionalized compounds. Consequently, a number of reactions involving oxygen removal (e.g., dehydration, hydrogenation, and hydrogenolysis), combined with C–C coupling (e.g., aldol condensation, ketonization, and oligomerization), will be required to convert sugars into hydrocarbon transportation fuels, and aqueous-phase catalytic processing offers the opportunity to selectively carry out those transformations. Importantly, two aspects are crucial to ensure economic feasibility of the aqueous-phase route: (i) reduction of the number of processing steps by means of catalytic coupling approaches92 and (ii) deoxygenation of biomass feedstocks with minimal consumption of hydrogen from an external source.93
O and –COOH groups) and a maximum number of carbon atoms limited to six (derived from glucose monomers). On the other hand, hydrocarbon fuels are larger (up to C20 for diesel applications) and completely unfunctionalized compounds. Consequently, a number of reactions involving oxygen removal (e.g., dehydration, hydrogenation, and hydrogenolysis), combined with C–C coupling (e.g., aldol condensation, ketonization, and oligomerization), will be required to convert sugars into hydrocarbon transportation fuels, and aqueous-phase catalytic processing offers the opportunity to selectively carry out those transformations. Importantly, two aspects are crucial to ensure economic feasibility of the aqueous-phase route: (i) reduction of the number of processing steps by means of catalytic coupling approaches92 and (ii) deoxygenation of biomass feedstocks with minimal consumption of hydrogen from an external source.93
The main aqueous-phase routes to upgrade sugars and derivatives into liquid hydrocarbon transportation fuels are schematically shown in Fig. 6. The biomass derivatives have been selected in view of their potential to produce liquid hydrocarbon fuels. First, we will describe the catalytic route designed to convert glycerol into liquid hydrocarbon fuels. This route involves the integration of two processes: aqueous-phase reforming (APR) of glycerol to syngas and F–T synthesis. This approach is particularly interesting because glycerol is produced in large amounts as a waste stream of the growing biodiesel industry.94 Furthermore, glycerol can be co-produced, along with ethanol, by bacterial fermentation of sugars95 (Fig. 4). Secondly, we will address furfural and hydroxymethylfurfural (HMF) as important compounds obtained by chemical dehydration of biomass-derived sugars. Furfural and HMF can be used as platform chemicals for green diesel and jet fuel production through dehydration, hydrogenation and aldol-condensation reactions. More recently, our group has developed a two-step (involving sugar reforming/reduction and C–C coupling processes) cascade catalytic approach to convert aqueous solutions of sugars and polyols into the full range of liquid hydrocarbon fuels, and this process will be described in Section 4.3.3. Finally, we will analyze the potential of two important biomass derivatives, levulinic acid (LA, obtained from sugars or HMF through dehydration processes) and γ-valerolactone (GVL, obtained by hydrogenation of LA), to produce liquid hydrocarbon fuels.
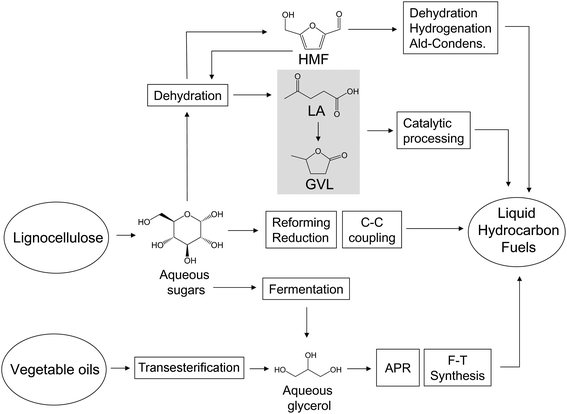 | ||
| Fig. 6 Main catalytic routes for the aqueous-phase conversion of sugars and derivatives into liquid hydrocarbon transportation fuels. | ||
To allow for coupling of endothermic glycerol reforming with exothermic Fischer–Tropsch synthesis in a single reactor, it is crucial that the former process is carried out efficiently at low temperatures. However, under these conditions, the metallic surface of Pt is likely to be saturated with adsorbed CO,101 thus decreasing the overall catalytic rate. One effective strategy to overcome this limitation involves the utilization of alloys such as Pt–Re and Pt–Ru, on which the strength of adsorption of CO is lower compared to Pt.102,103 This new route involving low-temperature gasification of aqueous glycerol to syngas integrated with Fischer–Tropsch synthesis represents an interesting alternative to complex BTL approaches (Section 4.1). Thus, unlike biomass gasification, glycerol reforming can be carried out at temperatures within the range employed for Fischer–Tropsch synthesis, thereby allowing effective integration of both processes in a single reactor104 with improved thermal efficiency (since heat required for endothermic reforming is provided by exothermic F–T process). Furthermore, concentrated aqueous solutions of glycerol (as produced in biodiesel facilities) can be converted to undiluted and impurity-free syngas, thereby eliminating the need for large gasifiers with oxygen-production plants and expensive gas-cleaning units. Thus, unlike BTL, this aqueous-phase route allows for cost-competitive operations at small scale, which is advantageous for the processing of distributed biomass resources.
Furfural and HMF can be used as building blocks for the production of linear hydrocarbons (in the molecular weight appropriate for diesel and jet fuel) by means of a cascade process involving dehydration, hydrogenation and aldol-condensation reactions,109,110 as shown in Fig. 7 for HMF. The process starts with acid-catalyzed deconstruction of polysaccharides (e.g., starch, cellulose or hemicellulose) to yield C5 and C6 sugar monomers, which are subsequently dehydrated (under the same acid environment) to form carbonyl-containing furan compounds such as furfural and HMF. In a further step, the carbonyl group in the furan compounds serves as a reactive center for C–C coupling through aldol condensation reactions with carbonyl-containing molecules such as acetone, which can be also obtained from biomass-derived sources.111,112 These condensations are base-catalyzed (e.g., NaOH, Mg–Al oxides) and are typically carried out in polar solvents like water. As a result of the aldol-condensation, a larger compound containing unsaturated CC and CO bonds (i.e., aldol-adduct) is formed and, owing to its hydrophobic character, this adduct precipitates out of the aqueous solution. Recently, improvements have been made in the aldol-condensation process by utilizing biphasic reactors where furan compounds (dissolved in organic THF) are contacted with aqueous NaOH, thus allowing continuous extraction of aldol-adducts into the organic phase.110 As represented in Fig. 7, the molecular weight of final alkanes can be increased by allowing adducts to undergo a second aldol-condensation process with the initial furanic feedstock. The unsaturated CC and CO bonds in aldol adducts are subsequently hydrogenated over metal catalysts such as Pd to yield large water-soluble polyol compounds. The complexity of the process can be reduced by using a bifunctional (metal and basic sites) water-stable Pd/MgO–ZrO2 catalyst.113 Thus, both aldol-condensation and adduct hydrogenation can be carried out simultaneously in a single reactor. The last step of the process involves complete oxygen removal from the hydrogenated aldol-adducts to produce liquid alkanes through aqueous-phase dehydration/hydrogenation (APD/H) reactions.114 Oxygen is progressively removed from the water-soluble adducts over a bifunctional metal–acid catalyst by cycles of dehydration and hydrogenation reactions. APD/H can be achieved over Pt–SiO2–Al2O3 in a four-phase reactor involving aqueous solution of adducts, a hydrogen gas inlet stream, a hexadecane sweep stream, and the solid catalyst.109 The hexadecane stream is important in that it prevents intermediate organic species from overreacting to coke over acid sites. Recently, the utilization of a bifunctional Pt/NbPO4 catalyst, with superior dehydration activity under water environments,115 has allowed elimination of the hexadecane sweep stream step and, consequently, production of a pure organic stream of liquid hydrocarbon fuels with targeted molecular weights (C9–C15 for HMF and C8–C13 for furfural) that spontaneously separates from water and retains 60% of the carbon of the initial sugar feedstock.110
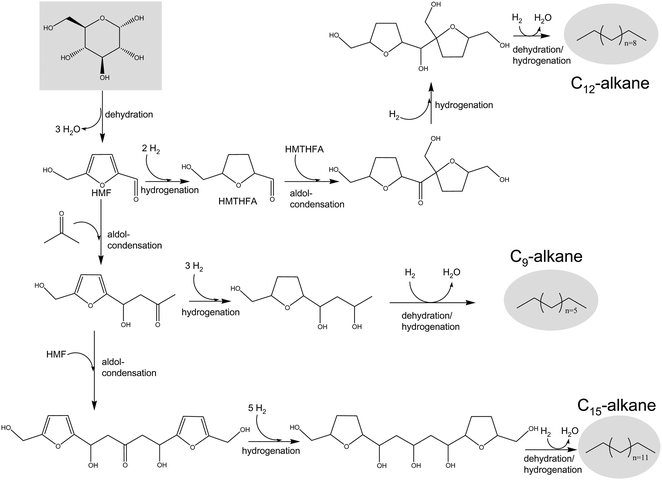 | ||
| Fig. 7 Reaction pathways for the conversion of biomass-derived glucose into liquid alkanes via HMF. Adapted from ref. 109. | ||
Both approaches are combined by Kunkes et al. in a recent process that transforms aqueous solutions of sugars and sugar-alcohols into liquid hydrocarbon fuels in a two-step cascade process116 (Fig. 8). Firstly, aqueous sugars and polyols (typically glucose and sorbitol) are converted into a mixture of monofunctional compounds (e.g., acids, alcohols, ketones and heterocycles) in the C4–C6 range, which are stored in an organic phase that spontaneously separates from water. This step is carried out at temperatures near 500 K over a Pt–Re/C catalyst, which achieves deep deoxygenation (up to 80% of the oxygen in the initial feedstock is removed) by means of C–O hydrogenolysis reactions. Importantly, the hydrogen required to accomplish the C–O cleavage step is internally supplied by aqueous-phase reforming (involving C–C cleavage and WGS reactions) of a fraction of the feed (Fig. 8). The Pt–Re/C material allows production of hydrogen and removal of oxygen in a single reactor. Unlike bio-oils produced by pyrolysis (Section 4.2), the organic stream of monofunctional compounds produced by sugar processing over Pt–Re/C is completely free of water and has a well-defined composition that is controlled by the feedstock type (e.g., sugars or polyols) and the reaction conditions.117
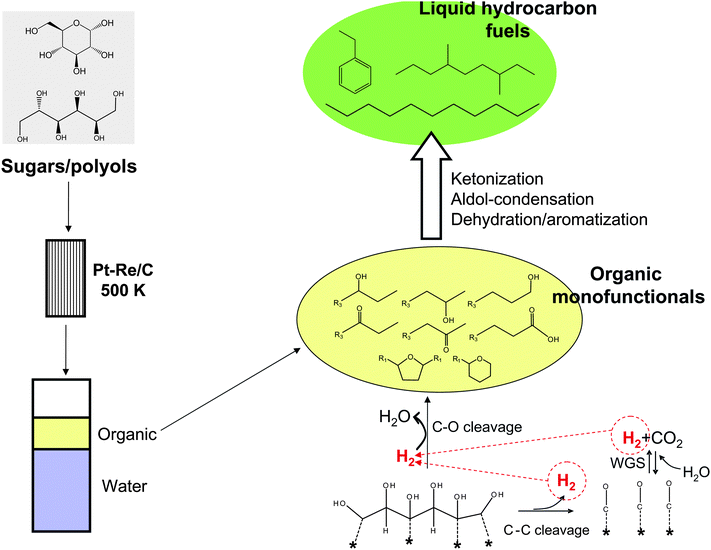 | ||
| Fig. 8 Scheme of the process for the catalytic conversion of sugars and polyols into liquid hydrocarbon fuels. Sugars primarily undergo reforming/reduction over Pt–Re/C to generate intermediate hydrophobic monofunctionals. The intermediates can be upgraded to liquid hydrocarbon fuels by means of C–C coupling reactions. Adapted from ref. 116. | ||
The retention of functionality in the organic intermediates is key to control reactivity and to allow subsequent C–C coupling upgrading strategies. This approach has been demonstrated to be conceptually adequate to process sugars into fuels,3 and important biomass derivatives such as lactic acid (3-hydroxypropanoic acid)89,118 and levulinic acid119 have been upgraded following this strategy. Each group of compounds (e.g., alcohols, ketones, acids) in the monofunctional stream can be upgraded to targeted hydrocarbons through different C–C coupling reactions (e.g., oligomerization, aldol-condensation and ketonization). For example, the organic stream enriched in alcohols by hydrogenation of ketones can be processed over an acidic H-ZSM5 zeolite at atmospheric pressure to yield 40% of C6+ aromatic gasoline components. Ketones can be upgraded to larger hydrocarbon compounds (C8–C12) with low extents of branching by means of aldol-condensation reactions over bifunctional Cu/Mg10Al7Ox catalysts. However, carboxylic acids present in the organic stream caused deactivation of the basic sites responsible for aldol-condensation, and approaches based on upstream removal of acids by ketonization (similar to those proposed herein for bio-oils upgrading, Section 4.2) and subsequent aldol-condensation have been successfully developed.120–122 Ketonization acquires special relevance when the organic stream is rich in carboxylic acids, as is the case when the feed is glucose.
Recently, our group has developed a series of catalytic approaches to convert aqueous solutions of levulinic acid into liquid hydrocarbon transportation fuels of different classes (Fig. 9). The catalytic pathways involve oxygen removal by dehydration/hydrogenation (in the form of water) and decarboxylation (in the form of CO2) reactions, combined with C–C coupling processes such as ketonization, isomerization, and oligomerization that are required to increase the molecular weight and to adjust the structure of the final hydrocarbon product. As a first step, aqueous levulinic acid is hydrogenated to water-soluble GVL, which is the key intermediate for the production of hydrocarbon fuels. This hydrogenation step can be achieved with high yields by operating at low temperatures (e.g., 423 K) over non-acidic catalysts (e.g., Ru/C) to avoid formation of angelica lactone, a known coke precursor125 which is produced by dehydration over acidic sites at higher temperatures (e.g., 573–623 K).119 Interestingly, because equimolar amounts of formic acid (a hydrogen donor) are co-produced along with levulinic acid in the C6-sugars dehydration process, this hydrogenation step could be potentially carried out without utilizing hydrogen from an external source, and several groups have already explored this possibility.126,127 This route is promising in that GVL has applications as a gasoline additive,128 and as a precursor to polymers129 and fine chemicals.130
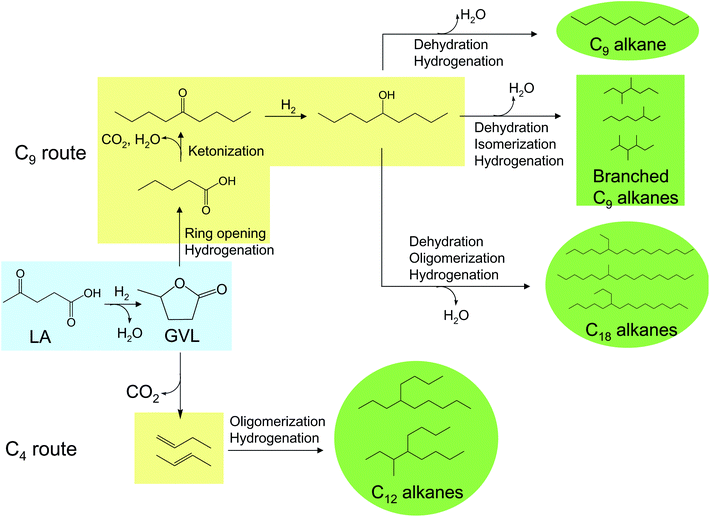 | ||
| Fig. 9 Catalytic routes for the conversion of levulinic acid (LA) and γ-valerolactone (GVL) into liquid hydrocarbon transportation fuels. Blue colour indicates water-soluble compounds, yellow symbolizes hydrophobic compounds, and green refers to liquid hydrocarbon fuels. | ||
Aqueous solutions of GVL can be upgraded to liquid hydrocarbon fuels by following two main pathways: the C9 route and the C4 route (Fig. 9). In the former route, GVL is converted to 5-nonanone over a water-stable multifunctional Pd/Nb2O5 catalyst. In this process, GVL is first transformed into hydrophobic pentanoic acid by means of ring-opening (on acid sites) and hydrogenation reactions (on metal sites) at moderate temperatures and pressures. Pentanoic acid is subsequently ketonized to 5-nonanone, and reaction conditions can be adjusted to allow this transformation to take place on the same Pd/Nb2O5 reactor with a maximum of 70% carbon yield.119 Nonanone yield can be increased to almost 90% by using a dual-catalyst approach with Pd/Nb2O5 + Ce0.5Zr0.5O2 in a reactor with two different temperature zones, which allows for optimum control of reactivity.131 5-Nonanone, which is obtained in a high purity organic stream that spontaneously separates from water, is subsequently transformed into its corresponding alcohol that serves as a platform molecule for the production of hydrocarbon fuels for gasoline and diesel applications. For example, the C9 alcohol can be processed (through hydrogenation/dehydration cycles) over a bifunctional metal–acid catalyst such as Pt/Nb2O5110 into linear n-nonane, with excellent cetane number and lubricity to be used as a diesel blender agent. Alternatively, the functionality of 5-nonanol can be utilized to upgrade the alcohol to gasoline and diesel components. In particular, 5-nonanol can be dehydrated and isomerized in a single step over an USY zeolite catalyst to produce a mixture of branched C9 alkenes with the appropriate molecular weight and structure for use in gasoline after hydrogenation to the corresponding alkanes.131 Additionally, 5-nonanol can be converted into a C9-alkene stream (by means of dehydration reactions) which can be subsequently oligomerized over an acid catalyst such as Amberlyst 70 to achieve good yields of C18 alkanes (after hydrogenation) for diesel applications.132
Recently, a promising route to upgrade aqueous solutions of GVL into jet fuels through the formation of C4 alkenes has been developed by Bond et al.133 (Fig. 9). The process is based on a dual reactor system. In the first catalytic reactor the GVL feed undergoes decarboxylation at elevated pressures (e.g., 36 bars) over a silica/alumina catalyst, producing a gas stream composed of butene isomers and CO2. In a second reactor connected in series, the gaseous butene stream is passed over an acidic catalyst (H-ZSM5, Amberlyst 70) that achieves oligomerization of butene monomers, yielding a distribution of alkenes centred at C12. While CO2 does not affect the oligomerization process other than by dilution, water inhibits the acidic oligomerization catalyst (especially Amberlyst), and it has to be removed prior to the second reactor by using a gas–liquid separator operating at 36 bars and 373–398 K. The final optimized yield to C8+ alkenes reaches 75% when silica/alumina and Amberlyst 70 are used.
Since GVL can be potentially produced from levulinic acid with no external hydrogen requirements, this technology allows the production of liquid hydrocarbon alkanes from lignocellulose with minimal utilization of hydrogen (i.e., hydrogen is only used during the final alkene hydrogenation step). Furthermore, the process is potentially cost-competitive with petroleum-derived technologies, since only two reactors are required, operating in series and using non-precious metal-catalysts. Finally, a CO2 gas stream is produced with high purity and at high pressures, thereby permitting effective utilization of sequestration or capture technologies to mitigate greenhouse gas emissions.
5. Comparison of technologies for biomass conversion into liquid hydrocarbon fuels
Table 1 summarizes the main characteristics of the different technologies analyzed in the present perspective paper for the conversion of biomass into liquid hydrocarbon fuels. Various routes have been compared in terms of important parameters (e.g., pretreatment required, external chemicals and hydrogen requirements, reaction conditions, cleaning/separation steps, number of reactors, use of precious metal catalysts) involved in the processing from the initial lignocellulosic biomass to the final liquid hydrocarbon fuel. Additionally, technologies are compared in terms of the overall yield of LHF for each route, calculated according to recent data available in literature. The ability to use the entire organic matter in lignocellulose represents the main advantage of thermal routes (i.e., BTL and pyrolysis + upgrading) versus aqueous-phase approaches which, as indicated in Section 4.3, only can process the sugar fraction of lignocellulose (typically 60–80% depending on the source13). This constraint in aqueous-phase routes has important consequences for the overall process: (i) it limits the final yield to LHF and (ii) it negatively affects the economics since additional reactors for pretreatment/hydrolysis steps are required to solubilize the sugar feedstocks (Table 1). On the other hand, aqueous-phase processes are carried out at milder conditions compared to thermal routes which allows for better control of the chemistry and, with it, higher selectivities to targeted hydrocarbon fuels. This control over the chemistry in aqueous phase technologies has important implications on the cleaning/separation steps as well. Thus, unlike BTL and pyrolysis which produce intermediate fractions with high degrees of impurities that require deep cleaning and conditioning before the upgrading process, aqueous-phase routes achieve high-purity organic streams that spontaneously separate from water, with no further cleaning/conditioning steps required. The higher number of reactors employed in the upgrading process for aqueous-phase routes versus thermal routes is another important difference between both approaches. In some cases (for example reforming/reduction of sugars, HMF and GVL platforms), the use of additional upgrading reactors is justified by the production of a final liquid hydrocarbon fuel with well-defined characteristics of molecular weight and structure that could be directly used for gasoline, jet fuel and diesel applications (see Fig. 7–9). This control of the final hydrocarbon fuel is more difficult to achieve by BTL and pyrolysis, which produce a mixture of hydrocarbons with broad molecular weight distributions and low control of the final chemical structure. Consequently, thermal routes might need additional refining reactors to produce fuel-grade compounds.| Technology | |||||||
|---|---|---|---|---|---|---|---|
| Thermal routes | Aqueous-phase routes | ||||||
| BTL | Pyrolysis | Glycerol reforming | HMF platform | Reforming of sugars | GVL platform | ||
| C9 route | C4 route | ||||||
a [![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ] indicates that hydrolysis and dehydration can be carried out in the same reactor.
b 3 reactors if WGS syngas conditioning is required.
c Depending on the upgrading process required.
d According to ref. 134.
e Calculated as: [0.50–0.70 yield of bio-oil from lignocellulose in pyrolysis135] × [0.65 yield of LHF in HDO,136 or 0.30 yield of aromatics LHF in zeolite upgrading79].
f Calculated as: [0.20 content of oil in soybeans137] × [0.1 glycerol in biodiesel process] × [0.552 yield of alkanes104].
g Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.53–0.31 yield isomerization glucose to fructose138,139] × [0.69–0.58 yield of LHF from fructose110].
h Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.52 yield of organic C116] × [0.57 yield of C7+ ketones116].
i Yield to n-nonane (diesel blender). Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.45 yield to levulinic acid of biofine process] × [0.96 yield of levulinic acid to GVL119] × [0.80 yield of 5-nonanone from GVL119] × [1.00] hydrogenation of 5-nonanone to n-nonane.
j Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.45 yield to levulinic acid of biofine process] × [0.96 yield of levulinic acid to GVL119] × [0.78 yield of C8+ alkenes133] × [1.00] hydrogenation to final alkane product. ] indicates that hydrolysis and dehydration can be carried out in the same reactor.
b 3 reactors if WGS syngas conditioning is required.
c Depending on the upgrading process required.
d According to ref. 134.
e Calculated as: [0.50–0.70 yield of bio-oil from lignocellulose in pyrolysis135] × [0.65 yield of LHF in HDO,136 or 0.30 yield of aromatics LHF in zeolite upgrading79].
f Calculated as: [0.20 content of oil in soybeans137] × [0.1 glycerol in biodiesel process] × [0.552 yield of alkanes104].
g Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.53–0.31 yield isomerization glucose to fructose138,139] × [0.69–0.58 yield of LHF from fructose110].
h Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.52 yield of organic C116] × [0.57 yield of C7+ ketones116].
i Yield to n-nonane (diesel blender). Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.45 yield to levulinic acid of biofine process] × [0.96 yield of levulinic acid to GVL119] × [0.80 yield of 5-nonanone from GVL119] × [1.00] hydrogenation of 5-nonanone to n-nonane.
j Calculated as: [0.80–0.60 sugar content of lignocellulose] × [0.96 yield of hydrolysis enzymatic] × [0.45 yield to levulinic acid of biofine process] × [0.96 yield of levulinic acid to GVL119] × [0.78 yield of C8+ alkenes133] × [1.00] hydrogenation to final alkane product.
|
|||||||
| Use of entire lignocellulose | Yes | Yes | No | No | No | No | No |
| Pretreatment required | Drying, size reduction | Drying, size reduction | Oil extraction | Pretreatment [acid hydrolysis dehydration]a, isomerization | Pretreatment, hydrolysis | Pretreatment [acid hydrolysis dehydration]a, hydrogenation | Pretreatment [acid hydrolysis dehydration]a, hydrogenation |
| External chemicals required | Oxygen | Inert gas | Methanol | Acetone, mineral acid, extracting solvent | Enzymes/mineral acid | Mineral acid | Mineral acid |
| Reaction conditions | 1100–1500 K, 1–20 bar | 650–800 K, 1 bar | 498–548 K, 1–20 bar | 298–393 K, 1–55 bar | 483–523 K, 18–27 bar | 598–698 K, 14–35 bar | 443–648 K, 36 bar |
| Intermediate fraction | Syngas | Bio-oil | Syngas | Hydrogenated aldol-adduct | Monofunctionals organic stream | 5-Nonanone-enriched organic phase | Butene + CO2 |
| Cleaning/conditioning/separation | Deep cleaning, WGS | Char removal | Not required | Extraction with organic solvent | Not required (spontaneous separation) | Not required (spontaneous separation) | Water removal |
| Upgrading process | F–T | HDO or zeolite upgrading | F–T | APD/H | C–C coupling, dehydration, hydrogenation | Hydrogenation, C–C coupling, dehydration | Oligomerization, hydrogenation |
| Number of reactors | 2–3b | 2 | 2 | 4 | 4 | 5–7c | 4 |
| Precious metal catalysts involved | No | Yes | Yes | Yes | Yes | Yes | No |
| External H2 requirements | No | High with HDO, none with zeolite | No | Moderate | No | Moderate | Minimum |
| LHF yield (g fuel per g dry biomass) | 0.21d | 0.45–0.15e | 0.011f | 0.27–0.10g | 0.23–0.16h | 0.25–0.19i | 0.25–0.19j |
As indicated in previous sections, two parameters are important to assess the economic feasibility of aqueous-phase catalytic routes: the number of reactors and the use of external hydrogen. Thus, glycerol reforming, with only 2 reactors (reforming and F–T) and no hydrogen requirements (Table 1), would represent an interesting route. However, the final hydrocarbon yield (0.011 g of LHF per g of dry biomass), negatively affected by the low content of oils in the biomass source (20% in soybeans137), is low compared with other aqueous routes. With respect to these two parameters, reforming/reduction of sugars over Pt–Re is a promising route since no external hydrogen is required (hydrogen required for reduction of sugars and hydrogenation of the ketone formed by aldol-condensation or ketonization is internally supplied in sufficient amounts by aqueous-phase reforming of a fraction of the sugar, Fig. 8), and only 4 reactors (hydrolysis, reforming, C–C coupling and dehydration/hydrogenation) are needed to produce gasoline, diesel and jet fuel components from lignocellulosic biomass with an overall yield comparable to that of BTL (0.21 g of LHF per g of dry biomass). The main drawback of this approach is the high cost of the Pt–Re (10 wt%) reforming catalysts. The recently developed GVL platform to produce butene oligomers offers an attractive alternative with minimum external hydrogen utilization (required only during the final alkene hydrogenation step) and no precious metal catalysts, which should give this process a promising economic assessment. The HMF platform route can achieve good yields to LHF (with a maximum yield close to 0.3) at the expense of needing external chemicals such as acetone and organic solvents (typically produced from fossil fuels like petroleum) and moderate amounts of hydrogen to carry out APD/H. The GVL C9 route offers versatility to produce gasoline and C9–C27 diesel components with acceptable yields, but it would require multiple reactors to transform biomass into the final hydrocarbon fuel depending on the upgrading process used. Finally, we note that taking into consideration all the parameters as a whole, pyrolysis coupled with upgrading processes appears to be a promising route to convert lignocellulose into LHF with high yields and low complexity. While advances have been made recently in the pyrolysis step, challenges for this route are currently focused on the upgrading process, with particular emphasis on two crucial aspects: (i) designing strategies for the reduction of hydrogen consumption during HDO and (ii) development of hydrothermally stable catalysts (preferably without precious metals) with high resistance to sulfur and alkaline impurities typically present in bio-oils.
6. Conclusions
The production of liquid transportation fuels from renewable sources such as biomass is a promising route that can help to reduce our dependence on fossil fuels and to mitigate global warming effects. Ethanol, the most abundantly produced biofuel at the present time, suffers from low energy-density and compatibility issues with the existing transportation infrastructure, which is based on petroleum-derived liquid hydrocarbons. The current limitations of ethanol as a fuel can be overcome by developing cost-effective technologies that allow conversion of non-edible lignocellulosic biomass into liquid hydrocarbon fuels chemically identical to those currently used in the transportation sector. In this respect, several promising routes are currently being developed worldwide, including gasification of biomass to syngas coupled with F–T synthesis (BTL), pyrolysis integrated with bio-oils upgrading processes, and aqueous-phase catalytic processing of biomass-derived sugars and derivatives. BTL and pyrolysis-upgrading are thermochemical routes that allow utilization of all the organic matter in lignocellulose, and aqueous-phase processing is a catalytic route designed to operate with water-soluble sugars (and platform chemicals derived from them). While aqueous-phase routes require that the lignocellulosic biomass be subjected to pretreatment/hydrolysis steps, these routes offer the opportunity to selectively carry out a variety of reactions to achieve the deep chemical transformations and C–C coupling reactions required when converting sugars into liquid hydrocarbons. To be economically viable, these aqueous-phase routes should be carried out with a small number of reactors and with minimum utilization of external fossil fuel-based hydrogen sources, as illustrated in the examples presented in the present paper.Acknowledgements
The results from the University of Wisconsin reported here were supported in part by the US Department of Energy Office of Basic Energy Sciences, by the DOE Great Lakes Bioenergy Research Center (http://www.greatlakesbioenergy.org/), and by the Defense Advanced Research Project Agency.References
- EIA Annual Energy Review, 2009, http://www.eia.doe.gov/aer/pdf/aer.pdf, accessed September 2010.
- Eurostat, Statistical Aspects of the Energy Economy, 2008, issue number 55/2009, http://epp.eurostat.ec.europa.eu/cache/ITY_OFFPUB/KS-SF-09-055/EN/KS-SF-09-055-EN.PDF, accessed September 2010 Search PubMed.
- D. Simonetti and J. A. Dumesic, ChemSusChem, 2008, 1, 725 CrossRef CAS.
- Energy Information Administration, International Energy Outlook, 2009, http://www.eia.doe.gov/oiaf/ieo/pdf/0484%282009%29.pdf, accessed September 2010.
- EIA International Energy Outlook, 2010, http://www.eia.doe.gov/oiaf/ieo, accessed September 2010.
- BP Statistical Review of World Energy, 2010, http://bp.com/statisticalreview, accessed September 2010.
- UK Energy Research Centre, The Global Oil Depletion Report, 2009, http://www.ukerc.ac.uk/support/Global%20Oil%20Depletion, accessed September 2010 Search PubMed.
- Climate Change, Synthesis Report. Intergovernmental Panel on Climate Change, 2007, http://www.ipcc.ch/publications_and_data/ar4/syr/en/contents.html, accessed September 2010 Search PubMed.
- President Bush State on the Union Address, White House, 2007, http://usgovinfo.about.com/b/2007/01/23/bush-delivers-his-seventh-state-of-the-union-address.htm, accessed September 2010.
- Directive 2003/30/EC of the European Union Parliament, Official Journal of the European Union, 2003, http://ec.europa.eu/energy/res/legislation/doc/biofuels/en_final.pdf, accessed September 2010.
- F. Kreith and D. Y. Goswami, in Handbook of Energy Efficiency and Renewable Energy, CRC Press: Taylor and Francis Group, Boca Raton, FL, 2007 Search PubMed.
- M. Graziani and P. Fornasiero, in Renewable Resources and Renewable Energy: A Global Challenge, CRC Press: Taylor and Francis Group, Boca Raton, FL, 2007 Search PubMed.
- D. L. Klass, in Biomass for the Renewable Energy, Fuels and Chemicals, Academic press, London, 1998 Search PubMed.
- J. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610 RSC.
- US National Science Foundation, in Breaking the Chemical and Engineering Barriers to Lignocellulosic Biofuels: Next Generation Hydrocarbon Biorefineries, 2008, http://www.ecs.umass.edu/biofuels/Images/Roadmap2-08.pdf, accessed September 2010 Search PubMed.
- Worldwatch Institute Center for American Progress, in American Renewable Path to Energy Security, 2006, http://www.worldwatch.org/files/pdf/AmericanEnergy.pdf, accessed September 2010 Search PubMed.
- International Energy Report, Energy Technology Essentials, Biofuel Production, 2007, http://www.iea.org/techno/essentials2.pdf, accessed September 2010.
- Renewable Fuels Association, Ethanol Industry Outlook, 2010, http://www.ethanolrfa.org/page/-/objects/pdf/outlook/RFAoutlook2010_fin.pdf%3Fnocdn%3D1, accessed September 2010.
- Worldwatch institute center for american progress, in Biofuels for Transport, Earthscan, London, 2007 Search PubMed.
- M. A. Martin, New Biotechnol., 2010 DOI:10.1016/j.nbt.2010.06.010.
- Biomass Research and Development Board, National biofuels action plan, 2008, http://www1.eere.energy.gov/biomass/pdfs/nbap.pdf, accessed September 2010.
- EPA/DOE sponsored web site, http://www.fueleconomy.gov/feg/flextech.shtml, accessed September 2010.
- J. R. Regalbuto, Science, 2009, 325, 822 CrossRef.
- Public Law 110–140, Energy Independence and Security Act of 2007, 2007, http://energy.senate.gov/public/_files/getdoc1.pdf, accessed September 2010.
- US Environmental Protection Agency report, Water Phase Separation in Oxygenated Gasoline, http://www.epa.gov/oms/regs/fuels/rfg/waterphs.pdf, accessed September 2010.
- Astm D 4806, Standard Specification for Denatured Fuel Ethanol for Blending with Gasoline for Use as Automotive Spark Ignition Engine Fuel, ASTM International, West Conshohocken, PA, 2010, DOI:10.1520/D4806-10, http://www.astm.org/.
- H. Shapouri, J. A. Duffield and M. Wang, in The Energy Balance of Corn Ethanol: An Update (report no. 814, Office of the Chief Economist), US Department of Agriculture, 2002, http://www.transportation.anl.gov/pdfs/AF/265.pdf, accessed September 2010 Search PubMed.
- Chemical Potential Energy, http://physics.info/energy-chemical, accessed September 2010.
- C. N. Hamelinck, R. A. A. Suurs and A. P. C. Faaij, Biomass Bioenergy, 2005, 29, 114 CrossRef.
- R. K. Perrin, in Ethanol and Food Prices—Preliminary Assessment, Faculty Publications: Agricultural Economics, University of Nebraska, 2008, http://digitalcommons.unl.edu/ageconfacpub/49, accessed September 2010 Search PubMed.
- D. L. Klass, in Biomass for the Renewable Energy and Fuels, Encyclopedia of Energy, ed. C. J. Cleveland, Elsevier, London, 2004 Search PubMed.
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, in Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-ton Annual Supply, Oak Ridge National Laboratory, 2005, DOE/GO-102005–2135, http://feedstockreview.ornl.gov/pdf/billion_ton_vision.pdf, accessed September 2010 Search PubMed.
- J. P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39 Search PubMed.
- J. J. Bozell, Clean, 2008, 36, 641 Search PubMed.
- M. Stocker, Angew. Chem., Int. Ed., 2008, 47, 9200 CrossRef CAS.
- D. Martin-Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- US Department of Energy, Feedstock Composition Glossary, 2005, http://www1.eere.energy.gov/biomass/feedstock_glossary.html%23C, accessed September 2010.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713 CrossRef CAS.
- A. Carrol and C. Somerville, Annu. Rev. Plant Biol., 2009, 60, 165 CrossRef.
- R. Kumar, G. Mago, V. Balan and C. E. Wyman, Bioresour. Technol., 2009, 100, 3948 CrossRef CAS.
- P. Dürre, Biotechnol. J., 2007, 2, 1525 Search PubMed.
- T. C. Ezeji, N. Qureshi and H. P. Blaschek, Curr. Opin. Biotechnol., 2007, 18, 220 CrossRef CAS.
- A. Schirmer, M. A. Rude, X. Li, E. Popova and S. B. del Cardayre, Science, 2010, 329, 559 CrossRef CAS.
- R. Luque, Energy Environ. Sci., 2010, 3, 254 RSC.
- J. Han, H. Sun, H. Lou and X. Zheng, Green Chem., 2010, 12, 463 RSC.
- I. Kubickova, M. Snare, K. Eranen, P. Maki-Arvela and D. Y. Murzin, Catal. Today, 2005, 106, 197 CrossRef CAS.
- UOP/ENI EcofiningTM Process, http://www.uop.com/objects/UOP_ENI_Ecofining_Process.pdf, accessed September 2010.
- L. R. Lynd, C. Wyman, M. Laser, D. Johnson and R. Landucci, in Strategic Biorefinery Analysis: Analysis of Biorefineries, Technical Report, National Renewable Energy Laboratory, USA, 2002, http://www.nrel.gov/docs/fy06osti/35578.pdf, accessed September 2010 Search PubMed.
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS.
- B. Kavalov and S. D. Peteves, in European Commission Joint Research Centre. Status and Perspectives of Biomass-to-Liquid Fuels in the European Union, 2005, http://www.mangus.ro/pdf/Stadiul%20actual%20si%20perspectivele%20bio-combustibililor%20in%20Europa.pdf, accessed September 2010 Search PubMed.
- T. A. Milne, R. J. Evans and N. Abatzoglou, in Biomass Gasifier Tars: Their Nature, Formation and Conversion, Report No. NREL/TP-570–25357, National Renewable Energy Laboratory, 1998, http://www.nrel.gov/docs/fy99osti/25357.pdf, accessed September 2010 Search PubMed.
- H. Boerrigter and A. Van Der Drift, in Biosyngas: Description of R&D Trajectory Necessary to Reach Large-Scale Implementation of Renewable Syngas from Biomass, Energy Research Centre of the Netherlands, 2004 Search PubMed.
- P. L. Spath and D. C. Dayton, in Preliminary Screening-Technical and Economic Assessment of Synthesis Gas to Fuels and Chemicals with Emphasis on the Potential for Biomass-Derived Syngas, United States Department of Energy, National Renewable Energy Laboratory, 2003, http://www.nrel.gov/docs/fy04osti/34929.pdf, accessed September 2010 Search PubMed.
- L. Caldwell, in Selectivity in Fischer–Tropsch Synthesis: Review and Recommendations for Further Work, 1980, http://www.fischer-tropsch.org/DOE/DOE_reports/81223596/pb81223596.pdf, accessed September 2010 Search PubMed.
- M. E. Dry, Catal. Today, 2002, 71, 227 CrossRef CAS.
- A. Steynberg, Fischer–Tropsch Technology, Studies on Surface Science and Catalysis, 2004, vol. 152, p. 1 Search PubMed.
- J. P. Lange, Catal. Today, 2001, 64, 3 CrossRef CAS.
- R. Zhang, K. Cummer, A. Suby and R. C. Brown, Fuel Process. Technol., 2005, 86, 861 CrossRef CAS.
- S. Koppatz, C. Pfeifer, R. Rauch, H. Hofbauer, T. Marquard-Moellensted and M. Specht, Fuel Process. Technol., 2009, 90, 914 CrossRef CAS.
- D. C. Elliott, D. Beckman, A. V. Bridgwater, J. P. Diebold, S. B. Gevert and Y. Solantausta, Energy Fuels, 1991, 5, 399 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848 CrossRef CAS.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68 RSC.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590 CrossRef.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147 CrossRef CAS.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324 CrossRef CAS.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. Melián Cabrera, R. H. Venderbosch and H. J. Heeres, Energy Environ. Sci., 2010, 3, 962 RSC.
- S. R. A. Kersten, W. P. M. van Swaaij, L. Lefferts and K. Seshan, Options for Catalysis in the Thermochemical Conversion of Biomass into Fuels, in Catalysis for Renewables: From Feedstock to Energy Production, ed. G. Centi and R. A. van Santen, Wiley-VCH, Weinheim, 2007 Search PubMed.
- S. Czernik, R. French, C. Feik and E. Chornet, Ind. Eng. Chem. Res., 2002, 41, 4209 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS.
- R. R. Davda and J. A. Dumesic, Chem. Commun., 2004, 36 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412 RSC.
- K. Ramesh, N. Sharma and N. Bakhshi, Energy Fuels, 1993, 7, 306 CrossRef CAS.
- J. D. Adjaye, S. P. R. Katikameni and N. N. Bakhshi, Fuel Process. Technol., 1996, 48, 115 CrossRef.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610 CrossRef CAS.
- T. R. Carlson, T. P. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397 CrossRef CAS.
- M. Renz, Eur. J. Org. Chem., 2005, 979 CrossRef CAS.
- K. M. Dooley, A. K. Bhat, C. P. Plaisance and A. D. Roy, Appl. Catal., A, 2007, 320, 122 CrossRef CAS.
- T. S. Hendren and K. M. Dooley, Catal. Today, 2003, 85, 333 CrossRef CAS.
- C. A. Gartner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, J. Catal., 2009, 266, 71 CrossRef CAS.
- C. A. Gartner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027 CrossRef.
- T. A. Milne, F. Aglevor, M. S. Davis, D. Deutch, and D. Johnson, in Development in Thermal Biomass Conversion, ed. A. V. Bridgewater and D. G. B. Boocock, Blackie Academic and Professional, London, UK, 1997 Search PubMed.
- R. Klimkiewicz, E. Fabisz, I. Morawski, H. Grabowska and L. Syper, J. Chem. Technol. Biotechnol., 2001, 76, 35 CrossRef CAS.
- M. Glinski, W. Szymanski and D. Lomot, Appl. Catal., A, 2005, 281, 107 CrossRef CAS.
- C. A. Gartner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, ChemSusChem, 2009, 2, 1121 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2009, 11, 1101 RSC.
- T. Werpy and G. Petersen, in Top Value Added Chemicals from Biomass, Vol. 1—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, US Dep. Energy, Off. Sci. Tech. Inf., 2004, http://www.nrel.gov/docs/fy04osti/35523.pdf, accessed September 2010 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- D. A. Simonetti and J. A. Dumesic, Catal. Rev., 2009, 51, 441 CrossRef CAS.
- J. C. Serrano-Ruiz, R. M. West and J. A. Dumesic, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 79 Search PubMed.
- M. Pagliaro and M. Rossi, in Future of Glycerol, New Usages for a Versatile Raw Material, RSC publishing, Cambridge, 2nd edn, 2010 Search PubMed.
- C. S. Gong, J. X. Du, N. J. Gao and G. T. Tsao, Appl. Biochem. Biotechnol., 2000, 84, 543 CrossRef.
- D. Gulen, M. Lucas and P. Claus, Catal. Today, 2005, 102–103, 166 CrossRef.
- R. S. Karinen and A. O. I. Krause, Appl. Catal., A, 2006, 306, 128 CrossRef CAS.
- T. S. Wiinikainen, R. S. Karinen and A. O. I. Krause, Conversion of Glycerol into Traffic Fuels, in Catalysis for Renewables: From Feedstocks to Energy Production, ed. G. Centi and R. A. van Santen, Wiley-VCH, Weinheim, 2007 Search PubMed.
- R. R. Soares, D. A. Simonetti and J. A. Dumesic, Angew. Chem., Int. Ed., 2006, 45, 3982 CrossRef CAS.
- R. Alcala, M. Mavrikakis and J. A. Dumesic, J. Catal., 2003, 218, 178 CrossRef CAS.
- R. He, R. R. Davda and J. A. Dumesic, J. Phys. Chem. B, 2005, 109, 2810 CrossRef CAS.
- E. Christoffersen, P. Liu, A. Ruban, H. L. Skriver and J. K. Norskov, J. Catal., 2001, 199, 123 CrossRef CAS.
- J. Greely and M. Mavrikakis, Nat. Mater., 2004, 3, 810 CrossRef CAS.
- D. A. Simonetti, R. Hansen, E. L. Kunkes, R. R. Soares and J. A. Dumesic, Green Chem., 2007, 9, 1073 RSC.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many by-Products, in Sugar Series, Elsevier, Amsterdam, 1st edn, 2000, vol. 13, pp. 34–69 Search PubMed.
- J. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
- C. Moreau, M. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS.
- Y. Roman-Leshkov, J. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS.
- G. W. Huber, J. Chheda, C. Barret and J. A. Dumesic, Science, 2005, 308, 1447.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417 CrossRef CAS.
- D. T. Jones and D. R. Woods, Microbiol. Rev., 1986, 50, 484 CAS.
- M. Sasaki, K. Goto, K. Tajima, T. Adschiri and K. Arai, Green Chem., 2002, 4, 285 RSC.
- C. Barret, J. Chheda, G. W. Huber and J. A. Dumesic, Appl. Catal., B, 2006, 66, 111 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549 CrossRef CAS.
- D. Braden, R. West and J. A. Dumesic, J. Catal., 2009, 262, 134 CrossRef CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS.
- R. M. West, E. L. Kunkes, D. A. Simonetti and J. A. Dumesic, Catal. Today, 2009, 147, 115 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, ChemSusChem, 2009, 2, 581 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574 RSC.
- E. L. Kunkes, E. Gurbuz and J. A. Dumesic, J. Catal., 2009, 266, 236 CrossRef CAS.
- E. Gurbuz, E. L. Kunkes and J. A. Dumesic, Green Chem., 2010, 12, 223 RSC.
- E. Gurbuz, E. L. Kunkes and J. A. Dumesic, Appl. Catal., B, 2010, 94, 134 CrossRef.
- S. W. Fritzpatrick, World patent, 9640609, Biofine Incorporated, 1997.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang and G. G. Neuenscwander, Resour., Conserv. Recycl., 2000, 28, 227 CrossRef.
- P. Ayoub and J. P. Lange, World patent, WO/2008/142127, 2008.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247 RSC.
- L. Deng, J. Li, D. M. Lai, Y. Fu and Q. X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529 CrossRef CAS.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- J. P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488 RSC.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249 CrossRef CAS.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184 CrossRef CAS.
- D. Martin-Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992 RSC.
- J. Q. Bond, D. Martin-Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS.
- D. Bianchi, in Biomass Catalytic Conversion to Diesel Fuel: Industrial Experience, Next Generation Biofuels, Bologna, 18 September 2009, http://www.ics.trieste.it/media/139853/df6504.pdf, accessed October 2010 Search PubMed.
- Biomass Technology Group (BTG), Fast pyrolysis, http://www.btgworld.com/index.php%3Fid%3D22%26rid%3D8%26r%3Drd, accessed October 2010.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. Melian-Cabrera, R. H. Venderbosch and H. J. Heeres, Energy Environ. Sci., 2010, 3, 962 RSC.
- USDA Nutrient database, http://www.nal.usda.gov/fnic/foodcomp/cgi-bin/list_nut_edit.pl, accessed October 2010.
- M. Moliner, Y. Roman-Leshkov and B. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164 CrossRef CAS.
- W. Aehle, in Enzymes in Industry: Production and Applications, Wiley-VCH, Weinheim, 2nd edn, 2004, p. 198 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |