Absorption into fluorescence. A method to sense biologically relevant gas molecules
Maria
Strianese†
a,
Antonio
Varriale†
b,
Maria
Staiano
bc,
Claudio
Pellecchia
a and
Sabato
D'Auria
*b
aDepartment of Chemistry, University of Salerno, Via Ponte Don Melillo, Fisciano, Sa I84084, Italy
bLaboratory for Molecular Sensing, IBP-CNR, Via Pietro Castellino, 111, 80131, Naples, Italy. E-mail: s.dauria@ibp.cnr.it
cUniversity of Siena, Italy
First published on 9th November 2010
Abstract
In this work we present an innovative optical sensing methodology based on the use of biomolecules as molecular gating nano-systems. Here, as an example, we report on the detection of analytes related to climate change. In particular, we focused our attention on the detection of nitric oxide (NO) and oxygen (O2). Our methodology builds on the possibility of modulating the excitation intensity of a fluorescent probe used as a transducer and a sensor molecule whose absorption is strongly affected by the binding of an analyte of interest used as a filter. The two simple conditions that have to be fulfilled for the method to work are: (a) the absorption spectrum of the sensor placed inside the cuvette, and acting as the recognition element for the analyte of interest, should strongly change upon the binding of the analyte and (b) the fluorescence dye transducer should exhibit an excitation band which overlaps with one or more absorption bands of the sensor. The absorption band of the sensor affected by the binding of the specific analyte should overlap with the excitation band of the transducer. The high sensitivity of fluorescence detection combined with the use of proteins as highly selective sensors makes this method a powerful basis for the development of a new generation of analytical assays. Proof-of-principle results showing that cytochrome c peroxidase (CcP) for NO detection and myoglobin (Mb) for O2 detection can be successfully used by exploiting our new methodology are reported. The proposed technology can be easily expanded to the determination of different target analytes.
Introduction
Fuel combustion is the primary source of a large number of damaging air pollutants, including, among others, fine particulate matter, carbon monoxide, and oxides of nitrogen. Emissions of these gases can cause many local and global environmental problems such as acid rain, greenhouse effect, destruction of the ozone layer and air pollution known as “smog”.1–5 Thus, the requirement for detecting hazardous gases in a wide range of everyday applications and, in particular, in the field of environmental analysis is becoming of great significance.A different key issue in environmental analysis is the monitoring of oxygen levels. Nowadays it is a routine practice to detect oxygen levels continuously in the atmosphere and in water.6 To better understand dynamics and drivers of oxygen depletion there is a pressing need for methods to monitor oxygen continuously. Recently optofluidic devices where hemoglobin was incorporated into free-standing silk diffraction gratings have been reported as simple and efficient oxygen sensors.7 The unique biophysical features of silk provide options to generate biomaterial systems perfectly suited for implementation in optical devices.7
In the present work, a new methodology for gas sensing applications by means of optical spectroscopy is described. Its overall characteristics make it an attractive candidate in the construction of devices for environmental analysis. Among the whole family of environmentally relevant gases we focused on the monitoring of nitric oxide (NO) and oxygen (O2). The proposed method builds on the possibility of modulating the excitation intensity of a fluorescent probe used as a transducer and a sensor molecule whose absorption is strongly affected by the binding of an analyte of interest used as a filter. In this work we present the results obtained with two different pairs of sensor-transducers: (1) cytochrome c peroxidase from baker's yeast (CcP) paired with fluorescein for detection of NO and (2) myoglobin from horse skeletal muscle (Mb) paired with fluorescein for detection of oxygen. Importantly, the proposed technology can be easily expanded to the determination of different target analytes.
Materials and methods
Materials
CcP was prepared following literature procedures.8 The purity of the CcP was checked spectrophotometrically by measuring the ratio of the absorbance at 408 nm and 280 nm or at 408 nm and 308 nm and comparing them with literature values (A408/A280 = 1.28 or A408/A308 = 1.57).8 The peak maximum at 408 nm (ε = 98.0 mM−1 cm−1) was used for determination of CcP samples concentration.8Fluorescein was purchased from Invitrogen. Stock solutions of fluorescein were prepared by dissolving the powder in milliQ water. Myoglobin from horse skeletal muscle, NaNO2, FeSO4·7H2O and H2SO4 (37% solution) was bought from Sigma Aldrich. The extinction coefficient of 29.1 mM−1 cm−1 at neutral pH and 280 nm was used to quote metMb sample concentrations.9Nitric oxide saturated solutions
Saturated solutions of nitrogen monoxide were prepared by bubbling NO gas, produced in situ according to literature procedures.10 150 ml of an acidic solution of ferrous sulfate in milliQ water were carefully dropped on solid NaNO2 (15 g). Under vigorous stirring of the reaction mixture, gaseous NO is quickly produced. The NO produced in this way was bubbled via cannula through 5 ml of 100 mM potassium phosphate (pH 6.8) for periods of up to 1 h, resulting in an approximate concentration of 2 mM at 20 °C.11Oxygen saturated solutions
Saturated solutions of oxygen were prepared by bubbling O2 gas, through 5 ml of 100 mM potassium phosphate (pH 6.8) for periods of up to 1 h resulting in an approximate concentration of 1.3 mM at 20 °C.11Sensor preparation
In a typical experiment the sensor was prepared as follows. A commercially available nitrocellulose strip (Schleicher & Schuell BioScience) was uniformly covered with a milliQ water solution of the dye-label fluorescein. After drying, the strip was carefully fixed to one of the external windows of a 10 × 10 mm2 airtight quartz fluorescence cuvette (Hellma Benelux bv, Rijswijk, Netherlands). To fix the nitrocellulose strip a black tape was used. The black tape acted also as black screen of the device. Fluorescence emission of the black tape upon excitation at 450 nm was checked for avoiding eventual interferences with the signal of the sensor. After prolonged incubation of the labeled cuvette in a buffer solution (1–2 days), the buffer showed no trace of dye fluorescence or absorption. This suggests that when the dye-labeled strip is dried no leaking out of the label should occur. To assess the activity of the dye after drying the strip its fluorescence emission spectrum was registered upon excitation at 450 nm. No significant differences could be detected when comparing these fluorescence spectra to the ones registered with the fluorescein-dye in solution.The dye-labeled cuvette was then filled with either CcP or Mb (5–10 µM) in 100 mM potassium phosphate buffer (pH 6.8) and the measurement was started. A quenching of the initial fluorescence intensity was normally observed when adding the protein into the cuvette. The intensity of the quenching was dependent on the concentration of the protein in the cuvette and on the amount of fluorophore.
Absorbance and fluorescence measurements
Absorption spectra were recorded on a Cary-50 Spectrophotometer with a slit-width equivalent to a bandwidth of 5 nm. The fluorescence spectra were measured on a K2 fluorometer (ISS, Champaign, IL, USA) with excitation wavelength of 450 nm and the emission slit width of 1 nm. Measurements were performed in potassium phosphate buffer (100 mM, pH 6.8) at room temperature. High quality argon (<1 ppm O2) was used to deoxygenate the sample prior to each measurement and to displace NO. For the experiments requiring NO or O2 addition, the cuvette was filled with deoxygenated sample solutions, after which µl amounts of an NO-saturated or O2-saturated buffer were injected.Results and discussion
The proposed method uses a commercial cuvette filled with the sensing protein and whose external surface, downstream of the sensor on the light path, is uniformly covered with the fluorescence transducer (Fig. 1). The photo-detector reads the fluorescence intensity emission of the transducer. The reading is proportional to the amount of excitation light which in turn depends on the amount of the analyte bound to the protein.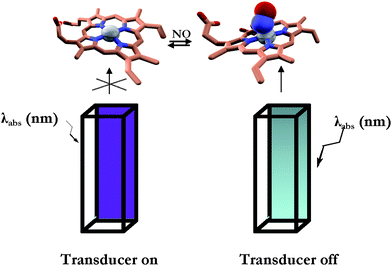 | ||
| Fig. 1 Schematic representation of the sensing methodology. Left: dye-labeled cuvette filled with CcP–NO free; right: dye-labeled cuvette filled with CcP–NO bound. See Materials and methods section for the experimental details. | ||
NO detection
CcP is a soluble 34 kDa heme protein, located in the inter-membrane space of yeast mitochondria.8 This protein is structurally, spectroscopically and functionally well characterized.8,12,13 Resting state CcP (FeIII) contains a non-covalently bound heme with a five-coordinate, high spin iron (S = 5/2).8 The sixth coordination position is vacant, allowing ligands like NO to bind. Several research groups have studied the interaction of CcP with small ligands other than NO (e.g., CN−, F−, CO).14,15 Some of the complexes formed upon interaction of CcP with small ligands were found not to be stable; for example the dioxygen adduct of CcP has a half life of 200 ms at 23 °C.16 By contrast, NO is one of the most powerful strong field ligands, which occurs on the right end of the spectrochemical series.17,18 One may expect, therefore, that the CcP–NO complex will exhibit excellent stability, which is clearly borne out when comparing, for example, the dissociation constant of the CcP–F− complex (305 µM)15 with that of the CcP–NO complex (10 µM19). Thus, the binding of NO to CcP is strongly preferred over other analytes.We have recently shown that fluorescently labeled CcP can be successfully used as a “turn-on” FRET-based biosensor for NO detection.19 We reported that when covalently attaching a proper fluorescent label to CcP and relying on Förster Resonant Energy Transfer between the label and the heme centre of the CcP, the fluorescent dye acts as a passive “beacon” which is “off” in the NO-free state and “on” in the NO-bound state of the protein.19 Drawing upon these findings, in the current work we explore the use of CcP as sensor to place inside the fluorescein-labeled cuvette and acting as the NO recognition element. In the present case no direct labeling of the CcP was performed. The fluorescence intensity of the dye-label is modulated by the different amounts of light absorbed by the CcP in the NO-free and NO-bound states. That is to say, the different absorption features of the CcP in the NO-free and NO-bound states change the amount of light reaching the dye transducer, thus tuning its fluorescence emission. Fig. 2 illustrates the absorption spectrum of CcP in the NO-free (black trace) and NO-bound (grey trace) forms.
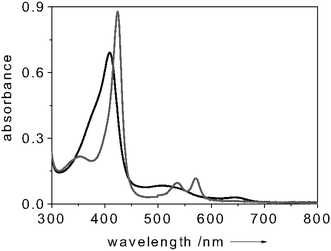 | ||
| Fig. 2 Absorption spectrum of NO-free (black) and NO-bound (grey) CcP. All spectra measured at room temperature. Protein concentration: 8 µM in 100 mM potassium phosphate buffer (pH = 6.8). | ||
In particular, upon NO binding to CcP, the Soret band shifts from 409 nm to 420 nm and its intensity increases. By choosing fluorescein (absorption maximum λ = 490 nm) as dye-transducer to functionalize the cuvette, this change in absorption upon NO binding can be translated into a change of fluorescence intensity of the label. To test the system, the fluorescence intensity of the dye-transducer (fluorescein, absorption maximum λ = 490 nm) was monitored during a change from an NO-free environment to an NO-saturated environment. Fig. 3 shows a typical fluorescence trace of a solution containing 5 µM of CcP when excited at 450 nm. When adding NO to the system (in considerable excess over the CcP concentration) a 20% decrease of the dye-label fluorescence was detected (Fig. 3). This finding is in accordance with the enhancement of the intensity of the Soret band of the CcP in the NO-bound form (Fig. 2) which leads the protein to absorb more photons. Subsequent bubbling of argon through the solution removed the NO again and made the fluorescence of the dye-label to go back to the initial level (Fig. 3). This showed that the NO binding process is reversible and our system works successfully when applied for monitoring NO. The CcP we are working with is in the Fe3+ state. Clear indications on the selectivity of the NO binding to CcP against O2 and CO are gained when considering that the Fe3+ form of the heme is unable to bind O2 and CO, as reported in the literature.16,20
 | ||
Fig. 3 Room temperature emission spectrum of CcP paired with fluorescein (exc 450 nm) (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) trace), upon addition (● trace) and removal (○ trace) of NO. Protein concentration: 5 µM in 100 mM potassium phosphate buffer (pH = 6.8). In this particular experiment the molar ratio NO to CcP was about 40 to 1. trace), upon addition (● trace) and removal (○ trace) of NO. Protein concentration: 5 µM in 100 mM potassium phosphate buffer (pH = 6.8). In this particular experiment the molar ratio NO to CcP was about 40 to 1. | ||
The application of heme proteins for NO detection is an already acknowledged finding. The hemoglobin-based assay has been widely used over the last decades.21 Differently from our method, this technique is based on the oxidation of reduced hemoglobin(Fe2+) by NO rather than on the detection of NO–Hb. NO is detected by observing the characteristic shift in the Soret absorbance peak of hemoglobin from 433 nm (hemoglobin(Fe2+)) to 406 nm (hemoglobin(Fe3+)), which means that the NO is measured in an indirect way.21
Oxygen detection
Myoglobin is a 18 kDa protein which is commonly acknowledged as an oxygen storage protein. It reacts with molecular oxygen by a simple association reaction, most likely due to its monomeric form.22–24Aiming at implementing an oxygen sensing device by exploiting the potential of our methodology, we considered myoglobin perfectly suited for proof-of-principle experiments. Since O2 has a strong preference for binding Mb in the Fe2+ state, in all our experiments we first reduced Mb to the Fe2+ form. Fig. 4 illustrates the absorption spectrum of Mb in the metMb(Fe3+), oxygen-free(Fe2+) and oxygen-bound(Fe2+) states.25–27
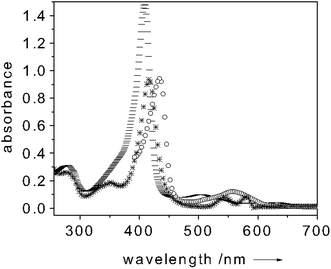 | ||
| Fig. 4
Absorption spectrum of metMb(Fe3+) ( trace), O2-free(Fe2+)Mb (○ trace) and O2-bound(Fe2+)Mb (* trace). All spectra measured at room temperature. Protein concentration: 10 µM in 100 mM potassium phosphate buffer (pH = 6.8). | ||
When adding a 3 mM sodium dithionite excess over the Mb concentration, a clear shift of the Soret band to 434 nm and the appearance of a band centered around 560 nm occur. These bands are diagnostics of the oxygen-free(Fe2+)Mb form.25–27 Upon oxygen binding the Soret band shifts again (to 417 nm) whereas the 560 nm band is quenched and two new absorption bands centered at 545 and 580 nm appear (Fig. 4).25–27 This change of the absorption spectrum strongly modulates the fluorescence intensity of the dye-transducer (fluorescein) used for functionalizing the cuvette. Fig. 5 shows a typical fluorescence trace of a solution containing 10 µM of Mb when excited at 450 nm. Upon Mb reduction to the oxygen-free(Fe2+) state, a 35% quenching of the dye-fluorescence is observed as a result of the fact that more light is absorbed by the Mb (see Fig. 5). As soon as oxygen binds Mb the fluorescence of the label increases (40%) (see Fig. 5). In the oxygen-bound state the fluorescein fluorescence is essentially uninhibited since the Soret band of Mb is quenched if compared to metMb(Fe3+) and shifts its maximum intensity to 417 nm (Fig. 4). When adding K3Fe(CN)6 (0.1 mM) the oxygen-bound Mb(Fe2+) turns to the initial metMb(Fe3+) and the initial fluorescence intensity of fluorescein is restored (see Fig. 5). It follows that the dye fluorescence is a sensitive reporter of the oxygen bound to the iron center. The cycle could be repeated many times. This finding showed that the O2 binding process is reversible, which is crucial for practical sensing applications. As for the selectivity of the O2 binding to Mb, the high affinity of Mb for the oxygen (Kd ≈ 1 µM) should ensure that the binding of this molecule is preferred over other analytes. On the other hand, the preliminary reduction of the metMb(Fe3+) to the oxygen-free(Fe2+) form, we perform in our measurements, constitutes a disadvantage for the selectivity of the O2 binding process to the Mb. As already mentioned above, also CO has a strong preference for binding Fe2+. Only changing the Mb for a protein with an affinity for O2 much higher than that for CO could ensure selectivity for O2 binding. Selectivity of neither the NO binding to CcP nor of the O2 to Mb could be checked against CN− due to the high shipping expenses of cyanide salts. (No changes in the fluorescence intensities were observed when different proteins (e.g. BSA) were used as controls.)
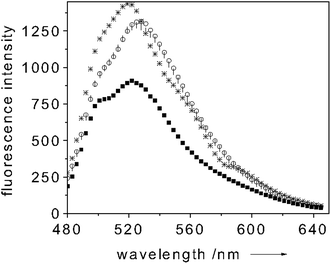 | ||
| Fig. 5 Room temperature emission spectrum of Mb paired with fluorescein (exc 450 nm) (○ trace), upon reduction to O2-free(Fe2+)Mb (■ trace), upon addition (* trace) and removal (| trace) of O2. Protein concentration: 10 µM in 100 mM potassium phosphate buffer (pH = 6.8). | ||
Concluding remarks
Our proposal is new in that it is based on a new principle, viz., the possibility of tuning the fluorescence emission of a dye-label (used to functionalize one of the window of a commercial fluorescence cuvette) by the different amounts of light absorbed by the protein which acts as sensing material. The method is applicable when the absorption spectrum of the sensing protein is strongly affected by the binding of the analyte of interest. The different absorption features of the analyte-free and analyte-bound protein states change the amount of light reaching the dye transducer, thus modulating its fluorescence intensities.A notable advantage of the new methodology we are proposing herein is its broad applicability. In principle it is a sensitive way to follow any event of interest given that it affects the absorption spectrum of the molecule acting as the recognition element.
Aiming at developing a commercial alternative to the current available environmental sensing devices, an additional feature would be that the affinity of the sensor for the analyte of interest follows in an appropriate range. Unfortunately, in our examples, the latter condition is not met, but we think it is important to examine the applicability of our method for the purpose of NO and O2 sensing as a proof-of-concept for our method.
Several examples of application can be found by simply choosing the proper pair sensor-transducer. Further experiments with this aim are currently underway in our lab. Just to mention a few examples: Mb paired with the commercial fluorescence-dye Cy3 as a H2S sensor. A redox protein (e.g. the small blue copper protein azurin paired with the Cy5 dye) to detect the protein redox state or, more generally, the redox potential of a solution while performing a reaction under specific conditions. Measurement of O2 consumption at sub-cellular levels and to monitor, for example, cell growth and viability or metabolic activity by using an oxygen sensing protein as molecular recognition element to place inside the cuvette are also under investigation.
Advantages like simplicity, rapidity in setting up the system and broad applicability make the developed device a possible alternative to the often time-consuming and expensive conventional screening assays used in the field of environmental analysis.
Differently from already existing fluorescence-based sensing devices, it avoids delaying steps due to the purification of the molecule acting as the recognition element upon fluorescent labeling.19,28–36
The dynamic range of the method can be adjusted not only by changing the concentrations of sensor and/or transducer but also by varying the dimension of the cuvette and the length of the light pathway.
Moreover, by using dye-transducers emitting at a specific wavelength the method permits us to exclusively monitor the event of interest even in complex mixtures.
Acknowledgements
This project was realized in the frame of the CNR Commessa “Diagnostica Avanzata ed Alimentazione” (SD, MS). The authors wish to thank Dr M. Baldassarre for the preparation of Fig. 1. SD wishes to thank Dr Paolo Bazzicalupo for the discussions and the constant support on the development of the bio-molecular gate concept.References
- G. Dooly, C. Fitzpatrick and E. Lewis, Energy, 2007, 33, 657–666.
- G. Dooly, C. Fitzpatrick and E. Lewis, J. Phys. Conf. Ser., 2007, 76, 012021 CrossRef.
- E. Hawe, G. Dooly, C. Fitzpatrick, P. Chambers, E. Lewis, W. Z. Zhao, T. Sun, K. T. V. Grattan, M. Degner, H. Ewald, S. Lochmann, G. Bramman, C. Wei, D. Hitchen, J. Lucas, A. Al-Shamma'a, E. Merlone-Borla, P. Faraldi and M. Pidria, Int. J. Intell. Syst. Tech. Appl., 2007, 3, 33–51 Search PubMed.
- S. Solomon, D. Qin, M. Manning, M. Marquis, K. Averyt, M. M. B. Tignor, H. LeRoy Miller, Jr, Z. Chen, Climate Change 2007; The Fourth Assessment Report (AR4) of the United Nations Intergovernmental Panel on Climate Change (IPCC), 2007.
- J. B. Smith, S. H. Schneider, M. Oppenheimer, G. W. Yohe, W. Hare, M. D. Mastrandrea, A. Patwardhan, I. Burton, J. Corfee-Morlot, C. H. Magadza, H. M. Fussel, A. B. Pittock, A. Rahman, A. Suarez and J. P. van Ypersele, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4133–4137 CrossRef CAS.
- Y. Amao, Microchim. Acta, 2003, 143, 1–12 CrossRef CAS.
- P. Domachuk, H. Perry, J. J. Amsden, D. L. Kaplan and F. G. Omenetto, Appl. Phys. Lett., 2009, 95, 253702 CrossRef.
- J. A. Worrall, U. Kolczak, G. W. Canters and M. Ubbink, Biochemistry, 2001, 40, 7069–7076 CrossRef CAS.
- L. Kelly and L. A. Holladay, Biochemistry, 1990, 29, 5062–5069 CrossRef CAS.
- B. Mattson, M. Anderson and S. Mattson, Microscale Gas Chemistry Book, Educational innovations, 4th edn, 2003 Search PubMed.
- G. W. C. Kaye and T. H. Laby, Tables of Physical and Chemical Constants, Longmans, London, 13th edn, 1966, p. 153 Search PubMed.
- T. Yonetani and H. Anni, J. Biol. Chem., 1987, 262, 9547–9554 CAS.
- A. E. Pond, M. Sono, E. A. Elenkova, D. E. McRee, D. B. Goodin, A. M. English and J. H. Dawson, J. Inorg. Biochem., 1999, 76, 165–174 CrossRef CAS.
- S. L. Edwards and T. L. Poulos, J. Biol. Chem., 1990, 265, 2588–2595 CAS.
- T. Yonetani and H. Anni, J. Biol. Chem., 1987, 262, 9547–9554 CAS.
- M. A. Miller, A. Shaw and J. Kraut, Nat. Struct. Biol., 1994, 1, 524–531 CrossRef CAS.
- R. Tsushida, Bull. Chem. Soc. Jpn., 1938, 13, 388–400 CAS.
- C. K. Jorgensen, Absorption Spectra and Chemical Bonding in Complexes, Pergamon Press, Oxford, 1962 Search PubMed.
- M. Strianese, M. F. De, V. Pavone, A. Lombardi, G. W. Canters and C. Pellecchia, J. Inorg. Biochem., 2010, 104, 619–624 CrossRef CAS.
- E. M. Boon and M. A. Marletta, J. Am. Chem. Soc., 2006, 128, 10022–10023 CrossRef CAS.
- S. Archer, FASEB J., 1993, 7, 349–360 CAS.
- M.-Y. R. Wang, B. M. Hoffman and S. J. Shire, J. Am. Chem. Soc., 1979, 101, 7394–7397 CrossRef CAS.
- K. D. Jurgens, S. Papadopoulos, T. Peters and G. Gros, News Physiol. Sci., 2000, 15, 269–274 Search PubMed.
- M. T. Wilson and B. J. Reeder, Exp. Physiol., 2008, 93, 128–132 Search PubMed.
- E. Chung Kwang, H. Lan Esther, S. Davidsson Michael, S. Dunn Bruce, S. Valentine Joan and I. Zink Jeffrey, Anal. Chem., 1995, 67, 1505–1509 CrossRef CAS.
- S. J. Millar, B. W. Moss and M. H. Stevenson, Meat Sci., 2010, 42, 272–288.
- I. Yamazaki, K. N. Yokota and K. Shikama, J. Biol. Chem., 1964, 239, 4151–4153 CAS.
- M. Strianese, G. Zauner, A. W. Tepper, L. Bubacco, E. Breukirk, T. J. Aartsma, G. W. Canters and L. C. Tabares, Anal. Biochem., 2009, 385, 242–248 CrossRef CAS.
- G. Zauner, M. Strianese, L. Bubacco, T. J. Aartsma, A. W. Tepper and G. W. Canters, Inorg. Chim. Acta, 2008, 361, 1116–1121 CrossRef CAS.
- S. Kuznetsova, G. Zauner, R. Schmauder, O. A. Mayboroda, A. M. Deelder, T. J. Aartsma and G. W. Canters, Anal. Biochem., 2006, 350, 52–60 CrossRef CAS.
- G. Zauner, E. Lonardi, L. Bubacco, T. J. Aartsma, G. W. Canters and A. W. Tepper, Chem.–Eur. J., 2007, 13, 7085–7090 CrossRef CAS.
- S. D'Auria and J. R. Lakowicz, Curr. Opin. Biotechnol., 2001, 12, 99–104 CrossRef CAS.
- M. Staiano, P. Bazzicalupo, M. Rossi and S. D'Auria, Mol. BioSyst., 2005, 1, 354–362 RSC.
- A. Varriale, M. Staiano, M. Rossi and S. D'Auria, Anal. Chem., 2007, 79, 5760–5762 CrossRef CAS.
- A. Varriale, M. Rossi, M. Staiano, E. Terpetschnig, B. Barbieri, M. Rossi and S. D'Auria, Anal. Chem., 2007, 79, 4687–4689 CrossRef CAS.
- M. Strianese, G. Zauner, A. W. Tepper, L. Bubacco, E. Breukink, T. J. Aartsma, G. W. Canters and L. C. Tabares, Anal. Biochem., 2009, 385, 242–248 CrossRef CAS.
Footnote |
| † MS and AV have equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2011 |