Medium-bridged lactams: a new class of non-planar amides
Michal
Szostak
and
Jeffrey
Aubé
*
Department of Medicinal Chemistry, University of Kansas, Structural Biology Center, 2121 Simons Drive, West Campus, Lawrence, KS 66047, USA. E-mail: jaube@ku.edu; Fax: +1 785.864.4496; Tel: +1 785.864.8179
First published on 11th November 2010
Abstract
Medium-bridged twisted lactams, in which a non-planar amide bond is achieved by incorporating the nitrogen atom at the bridgehead position in a medium-sized heterocycle, offer an attractive setting in which to study the properties of distorted amide linkages. This Emerging Area article will describe progress in the preparation and study of these compounds. This work shows that compounds containing an even moderately distorted amide bond display useful and unusual chemical properties while retaining a measure of stability that enables their study.
 Michal Szostak | Michal Szostak received his M.Sc. degree from Wroclaw Medical University, Poland, in 2005. He completed a Ph.D. at the University of Kansas in 2009 under the supervision of Professor Jeffrey Aubé, and is currently a postdoctoral researcher at Princeton University in the group of Professor David MacMillan. |
 Jeffrey Aubé | Jeff Aubé attended the University of Miami, where he did undergraduate research with Professor Robert Gawley. He received his Ph.D. in chemistry in 1984 from Duke University, working with Professor Steven Baldwin, and was an NIH postdoctoral fellow at Yale University with Professor Samuel Danishefsky. In 1986, he moved to the University of Kansas, where he is now Professor of Medicinal Chemistry. Professor Aubé’s research interests include organic synthesis and medicinal chemistry. |
Introduction
The amide bond is one of the most important functional groups in chemistry and biology. Amides link amino acids in peptides and proteins and are vital structural units in a myriad of synthetic polyamides, natural products, and pharmaceuticals. The properties of amide bonds result from the intervention of two major canonical structures, leading to a partial double bond character of the N–C(O) motif and manifest in a strong tendency of amide bonds to preserve planarity.1Nevertheless, not all amide bonds in peptides are perfectly planar.2 Moreover, N–C(O) rotation has long been proposed to occur in the enzymatic hydrolysis and cis-transisomerization of amide bonds.3 In addition, distorted amide bonds are key elements of β-lactam antibiotics as first encountered in fungi and subsequently refined by medicinal chemists.4 Deviation from planarity is accompanied by hybridization and geometry changes at nitrogen and leads to a host of modifications in chemical reactivity of amide bonds, only one of which is its hypersensitivity to hydrolysis (Fig. 1).5
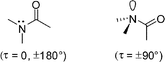 | ||
| Fig. 1 Planar and distorted amide bonds. | ||
All of this would seem to provide strong motivation for organic chemists to prepare and study amides with a range of degrees of twist. Indeed, some very important work has been registered in this area for over 70 years,6 but with an almost complete focus on highly twisted – and therefore extremely unstable – amides and the detailed study of only a single reaction (amide hydrolysis).1,7
We have begun to study amides that are substantially “twisted” thanks to their incorporation in particular medium-ring lactam scaffolds that permit the preparation and use of these compounds in a variety of settings, including aqueous solutions. In this article, we will argue that these compounds are readily prepared, display unconventional chemical properties, and represent a new direction for the study of non-planar amides.
Early work
Distortion of amide bonds can be achieved by geometric restriction,6 steric repulsion8a–8b or electronic delocalization,8c–8d however conformational restriction offers certain advantages over the latter methods (Fig. 2). Of these, geometrically restricted lactams are of special interest because they do not suffer from excessive steric hindrance around the amide bonds or from electronic influence of neighbouring substituents. Bridged lactam scaffolds can also be more easily modified and diversified, when compared to other classes of distorted amides.1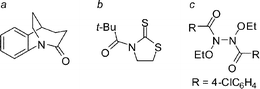 | ||
| Fig. 2 Examples of compounds in which amide bonds are distorted by: a) geometric restriction, b) steric repulsion, c) electronic delocalization. | ||
The first mention of a bridged lactam dates back to 1938 (the same time as the origin of Pauling's resonance theory). In that year, Lukeš proposed that incorporating a nitrogen at a bridgehead position in a bicyclic ring system would result in a violation of Bredt's rule by the amide zwitterionic resonance structure.9 Being unsuccessful in preparing some bridged amides by cyclizations (Scheme 1), Lukeš concluded that such amides are “sterically impossible” and if they were ever made they would exhibit properties of ketones rather than amides. In 1941, R. B. Woodward initiated a research program directed towards the problem of synthesizing the related [2.2.2] bicyclic 2-quinuclidone.10 Like Lukeš, Woodward appreciated that such compounds would represent a new breed of lactam structures.
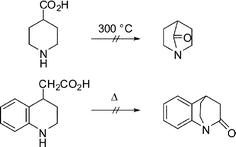 | ||
| Scheme 1 Attempted synthesis of anti-Bredt amides by Lukeš. | ||
Subsequent investigators had more success in preparing bridged bicyclic amides employing intramolecular condensations,11a–gcycloadditions11h and transition metal catalyzed11i–j approaches (Scheme 2). More recent work has expanded the palette to include bridged lactams containing larger ring systems and heteroatom-substituted versions (Fig. 3).12 However, these efforts usually generated a limited set of analogs and produced some compounds containing nearly planar amide linkages despite the bridged structures. In general, the follow up chemistry associated with these new compounds was not examined.
 | ||
| Scheme 2 Early examples of synthesis of bridged lactams. | ||
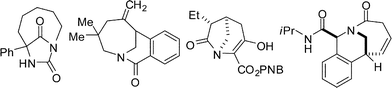 | ||
| Fig. 3 Selected examples of bridged lactams. | ||
In 1998, Kirby, while studying the reverse anomeric effect, achieved the synthesis of 1-aza-2-adamantanone, which he subsequently described as the “most twisted amide” (Scheme 3a).13 This nearly perfectly perpendicular amide (twist angle,14τ = 90.5°; sum of three bond angles at nitrogen, θ = 325.7°) displayed some very unusual keto-amine-like reactivity, including rapid hydrolysis in water (t1/2< 50 s), high basicity of the amide nitrogen (pKa ∼5.2) and spectroscopic properties typical of an amino ketone (IRνCO = 1732 cm−1, δ13C NMR = 200.0 ppm). In 2003, Coe utilized a homologue of 1-aza-2-adamantanone as an intermediate in the synthesis of nicotinic receptor ligands by subjecting it to an unprecedented Wolff–Kishner reduction (Scheme 3b).15 In both of these studies, the rigid adamantane-type structures facilitated the synthesis and influenced the properties of these lactams.
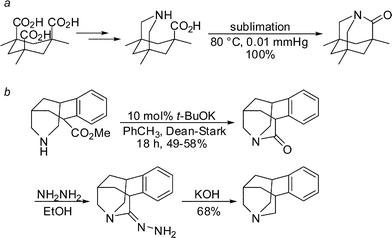 | ||
| Scheme 3 Synthesis of adamantane-derived bridged lactams. | ||
In 2006, Tani and Stoltz succeeded in synthesizing an iconic twisted amide, 2-quinuclidone (isolated as its tetrafluoroborate salt) using an intramolecular Schmidt ring expansion reaction (Scheme 4).16 This method allowed for the scrupulously anhydrous conditions required for isolation of this extremely unstable compound (in water, its t1/2 was less than 15 s). The X-ray structure of the protonated amide indicated a fully orthogonal amide bond (τ = 90.9° and pyramidalization at nitrogen,14χN = 59.5°).
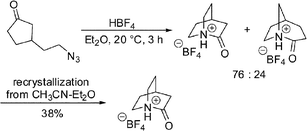 | ||
| Scheme 4 Synthesis of 2-quinuclidonium tetrafluoroborate. | ||
Prior to our studies (described in detail below) the potential of medium-bridged lactams could not be fully realized for the following reasons: (a) lack of unified synthetic approaches to medium-bridged lactams, (b) insufficient degree of distortion of amide bonds in lactams of this type that had been prepared (and thereby limiting new amide bond chemistry in these compounds) or (c) inadequate aqueous stability.
Discovery and preparation of medium-bridged lactams
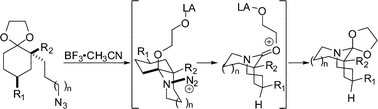
In 2002, we unexpectedly encountered a tricyclic bridged amide in the context of a total synthesis of stenine (3a, Scheme 5).17 In this domino reaction sequence, an intramolecular Diels–Alder reaction afforded an intermediate cis-decalone containing an axial azidoalkyl chain. The lactams were obtained by the subsequent C→N migration and loss of N2. Presuming migration of the bond antiperiplanar to the diazonium cation, the intermediate A containing the leaving group in a pseudoequatorial position led to the fused lactam whereas migration of the bond antiperiplanar to the N2+ in the pseudoaxial orientation afforded the previously unobserved bridged analogue. Importantly, the structure of tricyclic bridged amides was confirmed by X-ray crystallography, indicating that these compounds contain amide bonds from a previously unknown distortion range (τ = ca. 50°), and suggested some attractive possibilities for investigating the properties of compounds containing “half-way” rotated amide bonds.18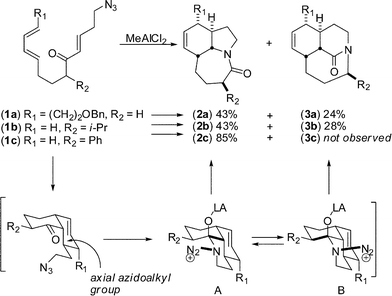 | ||
| Scheme 5 Synthesis of tricyclic planar and medium-bridged lactamsvia domino Diels–Alder/Schmidt sequence. | ||
In theory, when an azide-containing side chain is placed at the carbon adjacent to the electrophile, two constitutional isomers can be formed (Scheme 6).19 Formal insertion of the azide into the proximal C–C bond of the reactive electrophile would give a fused structure (path a), while the insertion into the distal C–C bond would give a bridged system (path b). The formation of 3a (Scheme 5) was completely unexpected insofar as all previous examples of the intramolecular Schmidt reaction had exclusively given fused amides.19b
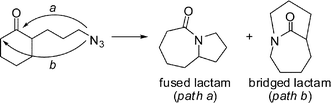 | ||
| Scheme 6 Potential regiochemical outcomes in the intramolecular Schmidt reaction of α-substituted ketones. | ||
We had previously proposed that chairlike cyclohexanone substrates can afford bridged products only when the azide-containing side chain and the diazonium cation occupy pseudoaxial orientations (Scheme 7, ax-tether, ax-cation).19a This is because other orientations – which are additionally favored for standard conformational reasons – do not permit an antiperiplanar relationship between the leaving group and the bond necessary for bridged lactam formation.
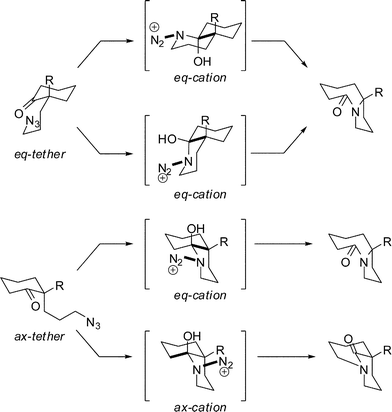 | ||
| Scheme 7 Reactive conformations in the intramolecular Schmidt reaction. | ||
We sought to expand these findings into a more general synthesis of bridged bi- and tricyclic lactams. After a few false starts, we proposed that an appropriately situated aromatic group might be able to stabilize an axial N2+ groupvia a through-space non-bonded cation–π interaction and accordingly persuade the desired bond to migrate. This was a risky proposition given that cation–π interactions are rarely invoked in organic chemistry and, to our knowledge, had not been previously used to influence the regiochemistry of a chemical reaction.20 However, we had previously deployed this idea in an asymmetric version of the Schmidt reaction21 and also considered the intriguing possibility that the same effect could be used to explain the complete formation of a fused lactam in the reaction of 1c→2c (cf.1b→2b + 3b, Scheme 5).22a
To test this hypothesis, a conformationally-locked α-phenyl substituted azide was prepared and found to afford a bridged lactam as the major product of its intramolecular Schmidt reaction (Table 1, entry 1).22a In this breakthrough discovery, which was the first time that the bridged product was observed to predominate against a fused possibility, the regiochemical control was achieved by the presence of both: (1) an axial tether and (2) the proposed cation–π interaction in the key azidohydrin intermediate (Table 1, box). Control reactions demonstrated that both the tert-butyl substituent and the aromatic ring were necessary for the efficient formation of the bridged products (entries 4–5). The observations that the bridged/fused ratio increased with a more electron-rich aromatic ring system and was found to be lower with an electron-withdrawing 4-nitrophenyl group provided strong support for the proposed cation–π interactions (entries 1–3).22b Subsequently, it was found that coordination of Lewis acids to substituents on the aromatic ring cannot be neglected in this system, emphasizing the importance of selecting appropriate reaction conditions to obtain maximum cation–π stabilization effects.
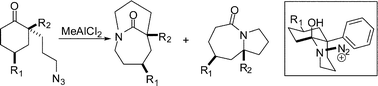
| Entry | R1 | R2 | Yield (%) | |
|---|---|---|---|---|
| Bridged | Fused | |||
| 1 | t-Bu | C6H5 | 61 | 22 |
| 2 | t-Bu | 4-(MeO)C6H4 | 71 | 11 |
| 3 | t-Bu | 4-(NO2)C6H4 | 39 | 38 |
| 4 | t-Bu | H | 17 | 57 |
| 5 | H | C6H5 | 0 | 96 |
This concept could be extended to substrates containing an α-heteroatomic group (Table 2).23 Importantly, the presumed cation–n stabilization operating in this system obviated the necessity for a locked conformation of the reactive azidohydrin intermediate. This not only significantly expanded the utility of Schmidt reaction for the synthesis of a range of one-carbon bridged lactams but also permitted the installation of a functional handle that could be subsequently converted to other useful chemotypes. The best results were obtained by placing thiomethyl substituent in the α-position (entries 1–3). From a theoretical viewpoint, these experiments have so far all been in agreement with our original ideas pertaining to the role of conformational effects in controlling the outcome of these Schmidt-like reactions (entries 4-5).19a The thiomethyl group was engaged in additional transformations to obtain a family of structurally related bridged lactams.
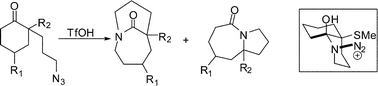
Interestingly, a significant increase in regioselectivity was noted when the Schmidt reaction was enacted on electrophilic species derived from ketals (Table 3).24Azide attack followed by rearrangement led to the formation of a relatively stable iminium ion intermediate, the intramolecular capture of which provided access to a set of unusual bridged orthoamides. These “α-amino ketals” are analogous to tetrahedral intermediates of amide addition reactions25 and can also be readily transformed into the parent twisted lactams.
Besides providing the first examples of any intramolecular Schmidt reaction affording exclusively bridged products, conformationally flexible 2-phenyl substituted azides cleanly furnished the bridged orthoamide (entry 5), while the [4.2.1] bridged scaffold was prepared employing a shorter azide chain (entry 6). It is likely that in the latter case, the fused product did not form due to strain developing en route to the corresponding four-membered-lactam containing fused product.26 The enhanced regioselectivity observed with ketal-containing vs.ketone-containing azides was ascribed to result from steric and electronic repulsion between the diazonium cation and the ketal tether. In addition, these reactions were not hampered by the necessity of forming a strained non-planar amide.
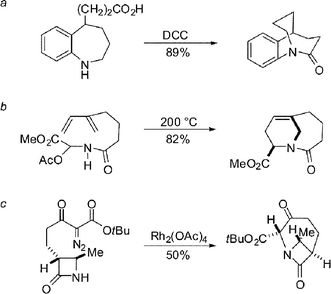
We thought it important to expand the availability of medium-bridged lactams to other ring systems. To accomplish this, we envisioned a new strategy based on the initial formation of a medium-sized ring, followed by a transannular cyclization reaction (Scheme 8).27 To this end, we found that ring-closing metathesis of simple diene precursors followed by transannular amidation provides a direct and straightforward access to a large family of medium-bridged lactams. This methodology was applied to the preparation of six different ring systems of one-carbon bridged lactams. Hydrogenation of double bonds provided access to unconstrained analogues. It should be noted that although synthesis of medium-sized nitrogen-containing heterocycles is challenging, the application of quaternary malonate esters facilitated these RCM reactions, permitting efficient preparation of the precursors for the final lactamization.
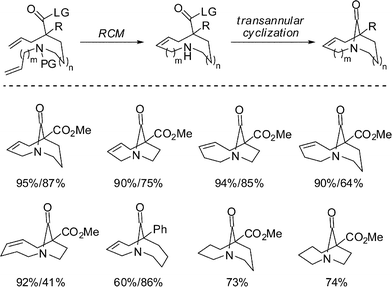 | ||
| Scheme 8 Direct two-step strategy for synthesis of bridged lactams. | ||
Importantly, although the medium-bridged lactams did not contain perfectly orthogonal amide bonds, these compounds exhibited more downfield shifts in the 13C NMR spectrum and higher stretching frequencies in the infrared spectrum corresponding to the amide bonds, which is consistent with a considerable degree of twist and ketone-like character of these amides (Table 4). Moreover, the infrared stretching frequencies of one-carbon bridged lactams covered a spectrum that starts in the range for planar amides (Table 4, entries 4–5), and ends close to that of a traditional ketone (entries 1–2), suggesting that the family of one-carbon bridged amides is well-suited for the systematic evaluation of the effect of geometry on properties of amide bonds.
Reactivity of bridged lactams
Due to the limited conjugation of amide bonds, distorted amides exhibit reactivity dissimilar to traditional amides. In general, the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O group is more electrophilic than in planar amides, while nitrogen, depending on the twist of the amide bond, might behave as a basic amine. At the outset of our studies, however, there were very few reports addressing twisted amide reactivity, with the vast majority of studies in place focused almost exclusively on amide bond hydrolysis.6 We thought that the intriguing reactivity profile of non-planar amide bonds deserved a broader look and began, like the pioneers before us, with hydrolysis chemistry.
O group is more electrophilic than in planar amides, while nitrogen, depending on the twist of the amide bond, might behave as a basic amine. At the outset of our studies, however, there were very few reports addressing twisted amide reactivity, with the vast majority of studies in place focused almost exclusively on amide bond hydrolysis.6 We thought that the intriguing reactivity profile of non-planar amide bonds deserved a broader look and began, like the pioneers before us, with hydrolysis chemistry.
Hydrolysis of medium-bridged amides
As a result of the enhanced electrophilicity of the amide carbonyl group, the vast majority of bridged amides is unstable to aqueous conditions. This hydrolytic instability complicates the synthesis and isolation of some of the more distorted bridged amides and is the single most important factor that has prevented a thorough investigation of the properties of these lactams (Fig. 4).1,28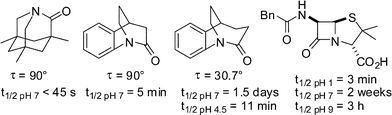 | ||
| Fig. 4 Hydrolytic instability of twisted and distorted amides. | ||
In sharp contrast, we determined that medium-bridged lactams are remarkably stable in aqueous solutions (Scheme 9).29 Their superior stability results from incorporation of the amide bond into a one-carbon bridge placed across a medium-sized heterocycle where it is subjected to scaffolding effects of medium-sized rings. Although we found small differences in the hydrolytic stability between tricyclic and bicyclic lactams, both types of structures are stable under biologically relevant pH conditions (pH = 4–10). This study culminated in the discovery of a bicyclic lactam that is not only stable but also fully water-soluble, which represents the first example of a significantly distorted bridged amide characterized by such properties. We note that these observations open up the possibility of bridged lactam-containing scaffolds in various biological applications.30
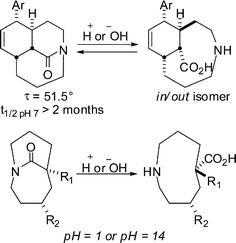 | ||
| Scheme 9 Hydrolytic behaviour of one-carbon bridged lactams. | ||
N-Activation of medium-bridged amides
Although oxygen is the protonation site of planar amide bonds, N-activation of amides has been invoked in discussions of various enzymatic processes and should at any rate provide access to new reactivity profiles of amides.1 Specifically, we expected the limited potential for nN→π*CO donation and the enhanced basicity of the nitrogen in medium-bridged lactams to engender potentially new reactivities of both theoretical and practical interest.
In fact, we experienced a surprise very early on in these studies when we discovered that one-carbon bridged amides undergo unprecedented C–N bond cleavage reactions under very mild reaction conditions (Scheme 10).18 Strikingly, the hydrogenation reactions were completely regioselective, with the C–N bond furthest away from co-planarity with the carbonyl group in the neutral lactam being selectively broken (Scheme 10a). It is likely that the amide bond nitrogen is activated for these reactions by forming a hydrogen bond with alcoholic solvents. Furthermore, tricyclic amides were found to undergo novel functionalization reactions on treatment with MeI and DDQ (Scheme 10b–c). These experiments fully established that distorted amides (and not only completely twisted lactams) display unique reactivity reaching beyond the known enhanced rate of hydrolysis or ketone-like reactions of amides. More recently, we found that the presence of an internal double bond increases reactivity of bicyclic lactams towards σ bond C–N hydrogenolysis.27,31
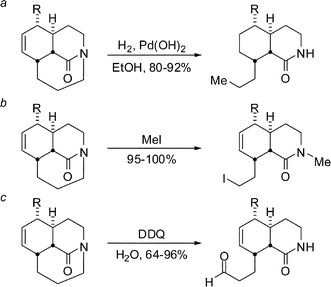 | ||
| Scheme 10 Regioselective cleavage of unactivated N–C bonds. | ||
Given the potential importance of N-protonation of distorted amides, we prepared several examples of N-protonated lactams and examined their structures by X-ray crystallography (Fig. 5).32 This study provided the first direct crystallographic comparison between any twisted amide and its corresponding salt. The ball-and-stick representations taken from the X-ray data clearly indicate a dramatic increase of pyramidalization around the C–N amide bond. We were unable to make the analogous salts from the moderately less distorted bicyclic analogs, which leads us to hypothesize that the twist/not-twist border from a reactivity perspective of amide bonds lies close to τ = 50°.
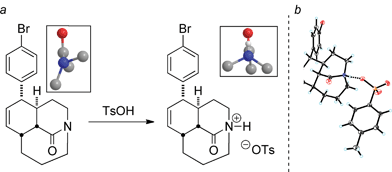 | ||
| Fig. 5 (a) N-Protonation of tricyclic bridged lactams (close-up of the amide region is shown in boxes, view along N–C(O) axis; ball-and-stick depictions of the X-ray derived structures). (b) Full crystal structure of N-protonated medium-bridged twisted amide. | ||
Synthesis of stable tetrahedral intermediates
The geometric constraints of non-planar amides should also greatly modify the chemistry of the affected carbonyl group and its derivatives. For example, Kirby reported the synthesis and X-ray structure of the N-protonated hydrate of 1-aza-2-adamantanone (Scheme 11).13b This compound may be viewed as a model for a cationic tetrahedral intermediate in acid-catalyzed acyl transfer reactions.25 The structure was characterized by a long C–N bond and a significantly shortened C–O bond, while its stability resulted from the torsional restriction imposed by the adamantane scaffold. Kirby had also reported the ketalization and Wittig methylenation of 1-aza-2-adamantanone,13a while related hydrazine formation was employed by Coe (Scheme 3).15 Aside from these examples, when we began our work, there was very little information available as to exactly how ketone-like twisted amides really were.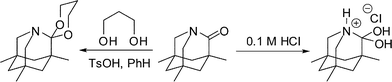 | ||
| Scheme 11 Tetrahedral intermediates from 1-aza-2-adamantanone. | ||
Conceptually, one might begin such a survey with simple nucleophilic addition reactions. Along these lines, we determined that medium-bridged lactams react with a mild reducing agent (NaBH4) and the resulting hemiaminals are stable to reaction and isolation conditions (Scheme 12a). When an electron-withdrawing group was α to the twisted amide bond, an unusual C–C bond cleavage reaction was observed (Scheme 12b).33
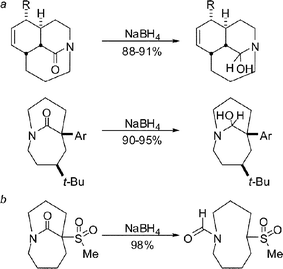
By subjecting our amides to organometallic additions of increasing steric demand (H− to t-BuLi), we learned that the resulting adducts undergo a transition from products having stable tetrahedral carbons to the collapsed ketone isomers (Scheme 13a). In some cases, intermediary products featuring intramolecular N⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions could be observed under certain conditions (Scheme 13b).33
O interactions could be observed under certain conditions (Scheme 13b).33
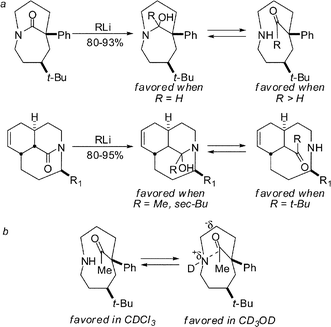 | ||
| Scheme 13 Transition from stable tetrahedral intermediates to ketone isomers in a series of medium-bridged twisted lactams. | ||
Kirby- and Coe-like formations of ketals and other common carbonyl derivatives were also observed (Scheme 14), solidifying support for the idea that even moderately distorted twisted amides really do behave like ketones.33
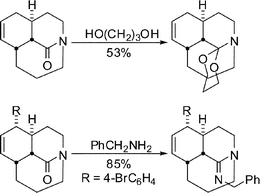 | ||
| Scheme 14 Synthesis of bridged aminoketal and amidine. | ||
The introduction of strain into the amide adducts would provide an even more severe test of the ability of the bridged amide to accommodate unusually stable “tetrahedral intermediates” of this type. Accordingly, we found that a family of aminoepoxides could be synthesized by direct Corey–Chaykovsky epoxidation of medium-bridged lactams with dimethylsulfonium methylide (Scheme 15).34 This transformation clearly benefits from the limited overlap of the lone pair of electrons of the amide nitrogen and the carbonyl systems. In strong analogy, we propose that the decreased nN-σ*C–O delocalization of the epoxide adducts is responsible for the stability of the resulting products.
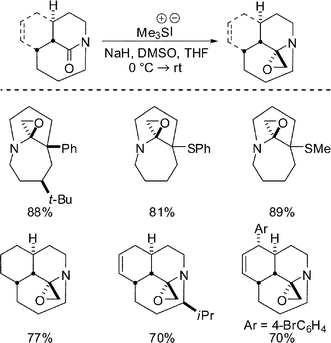 | ||
| Scheme 15 Synthesis of bridged aminoepoxides. | ||
We have begun to examine the reactivity of both α-amino ketals (formed in the Schmidt reaction of azido alkyl ketals, Table 3) (Scheme 16a)24 and epoxide adducts (Scheme 16b).34 These bridged heterocycles participate in a number of intriguing reactions including ring opening of the bicyclic system, N-protonation, and other rearrangements. The unusual reactivity patterns uncovered provide evidence that bridged systems of this type will accommodate bridged iminium ion intermediates and warrant further study.
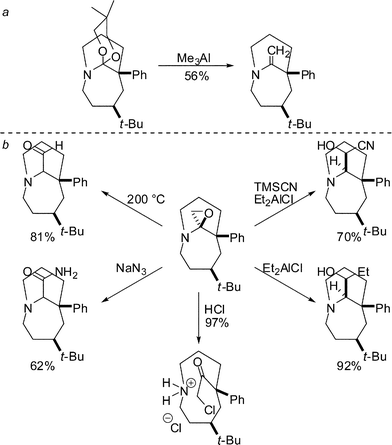 | ||
| Scheme 16 Reactivity of bridged aminoketals and aminoepoxides. | ||
Conclusions
Medium-bridged twisted lactams contain distorted amide linkages that, in contrast to traditional planar amide bonds, are vastly unexplored. The Schmidt reaction and transannular cyclization approach provide complementary routes to an unprecedented number of structurally related bridged amides. The use of electrostatic cation–π and cation–n interactions in the Schmidt rearrangement as a product-controlling feature is unusual and provides a potentially powerful new tool for stereoselective organic synthesis in general.The key advantages of one-carbon bridged lactams over other known models of distorted amides include enhanced stability in aqueous solutions, availability of systems with different geometries, and ease of structural diversification. Some intriguing results have already emerged from our initial studies. For example, reactions arising from N-activation of amide bonds, structural characterization of N-protonated lactams, and the synthesis of an array of previously unknown heterocycles have been described. Due to their aqueous stability profile, another potential application of medium-bridged lactams could be in drug discovery (i.e., as new scaffolds expanding available chemical space).30
Although non-planar amides and their properties have been of interest for the better part of a century, the discovery of medium-bridged twisted lactams constitutes a useful advance to the field by (1) showing that modification of amide bond properties does not require an all-or-nothing approach and (2) that this class of compounds has real potential for uses beyond the study of amide bond hydrolysis. We hope to contribute to this field through further study. In addition to finding useful applications of medium-bridged lactams, for example in total synthesis projects, our future work will be focused on gaining more insight into the quantitative evaluation of the distorted amide bonds contained in these fascinating molecules.
Acknowledgements
We thank the National Institute of General Medical Sciences (GM-49093) for financial support.Notes and references
- A. Greenberg, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000. For a detailed discussion of the types of distorted amide bonds, including a useful qualitative description, see: S. Yamada, in The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000, p. 215 Search PubMed.
- For some leading references, see: (a) G. N. Ramachandran, Biopolymers, 1968, 6, 1494 CrossRef CAS; (b) M. W. MacArthur and J. M. Thornton, J. Mol. Biol., 1996, 264, 1180 CrossRef CAS; (c) A. S. Edison, Nat. Struct. Biol., 2001, 8, 201 CrossRef CAS.
- For some leading references, see: (a) J. I. Mujika, J. M. Mercero and X. Lopez, J. Am. Chem. Soc., 2005, 127, 4445 CrossRef CAS; (b) G. Fischer, Chem. Soc. Rev., 2000, 29, 119 RSC.
- G. I. Georg, The Organic Chemistry of Beta-lactams, Wiley-VCH, 1992 Search PubMed.
- For some leading references, see: (a) A. Greenberg and C. A. Venanzi, J. Am. Chem. Soc., 1993, 115, 6951 CrossRef CAS; (b) J. I. Mujika, J. M. Matxain, L. A. Eriksson and X. Lopez, Chem.–Eur. J., 2006, 12, 7215 CrossRef CAS; (c) C. R. Kemnitz and M. J. Loewen, For an excellent study regarding calculation of the strain parameters of distorted amides in quinuclidone family, see:, J. Am. Chem. Soc., 2007, 129, 2521 CrossRef CAS; (d) A. Greenberg, D. T. Moore and T. D. DuBois, J. Am. Chem. Soc., 1996, 118, 8658 CrossRef CAS.
- For reviews, see: (a) H. K. Hall and A. El-Shekeil, Chem. Rev., 1983, 83, 549 CrossRef; (b) T. G. Lease, K. J. Shea, in Advances in Theoretically Interesting Molecules, JAI Press Inc., 1992, vol. 2, p. 79 Search PubMed; (c) J. Clayden and W. J. Moran, Angew. Chem., Int. Ed., 2006, 45, 7118 CrossRef CAS.
- For seminal contributions on quantitative evaluation of rates of hydrolysis of bridged lactams in quinuclidone series, see: (a) G. M. Blackburn, C. J. Skaife and I. T. Kay, J. Chem. Res. Synop., 1980, 294 Search PubMed; (b) A. J. Bennet, Q. P. Wang, H. Slebocka-Tilk, V. Somayaji, R. S. Brown and B. D. Santarsiero, J. Am. Chem. Soc., 1990, 112, 6383 CrossRef.
- (a) S. Yamada, Angew. Chem., Int. Ed. Engl., 1993, 32, 1083 CrossRef; (b) S. Yamada, Rev. Heteroat. Chem., 1999, 19, 203 Search PubMed; (c) A. M. E. Gillson, S. A. Glover, D. J. Tucker and P. Turner, Org. Biomol. Chem., 2003, 1, 3430 RSC; (d) S. Glover, Tetrahedron, 1998, 54, 7229 CrossRef CAS.
- R. Lukeš, Collect. Czech. Chem. Commun., 1938, 10, 148 CAS.
- (a) W. E. Doering and J. D. Chanley, J. Am. Chem. Soc., 1946, 68, 586 CrossRef CAS; See, also: (b) H. H. Wasserman, Nature, 2006, 441, 699 CrossRef CAS.
- For selected references, see: (a) N. F. Albertson, J. Am. Chem. Soc., 1950, 72, 2594 CrossRef CAS; (b) L. N. Yakhontov and M. V. Rubtsov, J. Gen. Chem. USSR, 1957, 27, 83 CAS; (c) H. Pracejus, Chem. Ber., 1959, 92, 988 CAS; (d) G. L. Buchanan, J. Chem. Soc., Chem. Commun., 1981, 814 RSC; (e) K. Steliou and M. A. Poupart, J. Am. Chem. Soc., 1983, 105, 7130 CrossRef CAS; (f) Q. P. Wang, A. J. Bennet, R. S. Brown and B. D. Santarsiero, J. Am. Chem. Soc., 1991, 113, 5757 CrossRef CAS; (g) A. Greenberg, G. L. Wu, J. C. Tsai and Y. Y. Chiu, Struct. Chem., 1993, 4, 127 CrossRef CAS; (h) T. G. Lease and K. J. Shea, J. Am. Chem. Soc., 1993, 115, 2248 CrossRef CAS; (i) R. Grigg, V. Sridharan, P. Stevenson and T. Worakun, J. Chem. Soc., Chem. Commun., 1986, 1697 RSC; (j) R. M. Williams, B. H. Lee, M. M. Miller and O. P. Anderson, J. Am. Chem. Soc., 1989, 111, 1073 CrossRef CAS.
- For selected references, see: (a) R. Grigg, V. Santhakumar, V. Sridharan, P. Stevenson, A. Teasdale, M. Thorntonpett and T. Worakun, Tetrahedron, 1991, 47, 9703 CrossRef CAS; (b) J. Aszodi, D. A. Rowlands, P. Mauvais, P. Collette, A. Bonnefoy and M. Lampilas, Bioorg. Med. Chem. Lett., 2004, 14, 2489 CAS; (c) T. P. Ribelin, A. S. Judd, I. Akritopoulou-Zanze, R. F. Henry, J. L. Cross, D. N. Whittern and S. W. Djuric, Org. Lett., 2007, 9, 5119 CrossRef CAS; (d) X. Coqueret, F. Bourellewargnier and J. Chuche, J. Org. Chem., 1985, 50, 909 CrossRef CAS; (e) W. J. Brouillette, V. P. Jestkov, M. L. Brown, M. S. Akhtar, T. M. Delorey and G. B. Brown, J. Med. Chem., 1994, 37, 3289 CrossRef CAS; (f) C. L. Molina, C. P. Chow and K. J. Shea, J. Org. Chem., 2007, 72, 6816 CrossRef CAS; For leading reference on structurally related bridged sultams, see: (g) L. A. Paquette and S. M. Leit, J. Am. Chem. Soc., 1999, 121, 8126 CrossRef CAS.
- (a) A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785 CrossRef CAS; (b) A. J. Kirby, I. V. Komarov and N. Feeder, J. Am. Chem. Soc., 1998, 120, 7101 CrossRef CAS; (c) A. J. Kirby, I. V. Komarov, P. D. Wothers, N. Feeder and P. G. Jones, Pure Appl. Chem., 1999, 71, 385 CrossRef CAS; (d) A. J. Kirby, I. V. Komarov and N. Feeder, J. Chem. Soc., Perkin Trans. 2, 2001, 522 RSC.
- F. K. Winkler and J. D. Dunitz, J. Mol. Biol., 1971, 59, 169 CrossRef CAS.
- C. G. Bashore, I. J. Samardjiev, J. Bordner and J. W. Coe, J. Am. Chem. Soc., 2003, 125, 3268 CrossRef CAS.
- K. Tani and B. M. Stoltz, Nature, 2006, 441, 731 CrossRef CAS.
- (a) J. E. Golden and J. Aubé, Angew. Chem., Int. Ed., 2002, 41, 4316 CrossRef CAS; (b) Y. B. Zeng and J. Aubé, J. Am. Chem. Soc., 2005, 127, 15712 CrossRef CAS; (c) K. J. Frankowski, J. E. Golden, Y. B. Zeng, Y. Lei and J. Aubé, J. Am. Chem. Soc., 2008, 130, 6018 CrossRef CAS.
- Y. Lei, A. D. Wrobleski, J. E. Golden, D. R. Powell and J. Aubé, J. Am. Chem. Soc., 2005, 127, 4552 CrossRef CAS.
- (a) G. L. Milligan, C. J. Mossman and J. Aubé, J. Am. Chem. Soc., 1995, 117, 10449 CrossRef CAS. For a recent comprehensive review on the intramolecular Schmidt reaction, see: (b) S. Grecian, J. Aubé, in Organic Azides: Syntheses and Applications, John Wiley and Sons, 2010, p. 191 Search PubMed; For insightful mechanistic discussion see, also: (c) W. H. Pearson, R. Walavalkar, J. M. Schkeryantz, W. K. Fang and J. D. Blickensdorf, J. Am. Chem. Soc., 1993, 115, 10183 CrossRef CAS.
- (a) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS; (b) S. Yamada, Org. Biomol. Chem., 2007, 5, 2903 RSC.
- (a) C. E. Katz and J. Aubé, J. Am. Chem. Soc., 2003, 125, 13948 CrossRef CAS; (b) C. E. Katz, T. Ribelin, D. Withrow, Y. Basseri, A. K. Manukyan, A. Bermudez, C. G. Nuera, V. W. Day, D. R. Powell, J. L. Poutsma and J. Aubé, J. Org. Chem., 2008, 73, 3318 CrossRef CAS; (c) T. Ribelin, C. E. Katz, D. G. English, S. Smith, A. K. Manukyan, V. W. Day, B. Neuenswander, J. L. Poutsma and J. Aubé, Angew. Chem., Int. Ed., 2008, 47, 6233 CrossRef CAS.
- (a) L. Yao and J. Aubé, J. Am. Chem. Soc., 2007, 129, 2766 CrossRef CAS; (b) M. Szostak, L. Yao and J. Aubé, J. Org. Chem., 2010, 75, 1235 CrossRef CAS.
- M. Szostak, L. Yao and J. Aubé, Org. Lett., 2009, 11, 4386 CrossRef CAS.
- M. Szostak and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2530 CrossRef CAS.
- M. Adler, S. Adler and G. Boche, J. Phys. Org. Chem., 2005, 18, 193 CrossRef CAS.
- This proposal was first outlined in the following references: (a) R. Iyengar, K. Schildknegt and J. Aubé, Org. Lett., 2000, 2, 1625 CrossRef CAS; (b) R. Iyengar, K. Schildknegt, M. Morton and J. Aubé, J. Org. Chem., 2005, 70, 10645 CrossRef CAS; During the preparation of this manuscript a report based on the above rationale was published: (c) F. Macleod, S. Lang and J. A. Murphy, Synlett, 2010, 529 CAS.
- M. Szostak and J. Aubé, Org. Lett., 2009, 11, 3878 CrossRef CAS.
- (a) V. Somayaji and R. S. Brown, J. Org. Chem., 1986, 51, 2676 CrossRef CAS; (b) M. A. Schwartz and F. H. Buckwalter, J. Pharm. Sci., 1962, 51, 1119 CrossRef CAS.
- M. Szostak, L. Yao and J. Aubé, J. Org. Chem., 2009, 74, 1869 CrossRef CAS.
- For pioneering application of bridged amides in medicinal chemistry, see: (a) E. E. Smissman, R. A. Robinson, J. B. Carr and A. J. Matuszak, J. Org. Chem., 1970, 35, 3821 CrossRef CAS; (b) W. J. Brouillette and H. M. Einspahr, J. Org. Chem., 1984, 49, 5113 CrossRef CAS; (c) W. J. Brouillette, G. L. Grunewald, G. B. Brown, T. M. DeLorey, M. S. Akhtar and G. Liang, J. Med. Chem., 1989, 32, 1577 CrossRef CAS.
- We also observed a similar enhanced reactivity of σ C–N amide bond in the synthesis of bridged thioamide: M. Szostak and J. Aubé, Chem. Commun., 2009, 7122 Search PubMed.
- M. Szostak, L. Yao, V. W. Day, D. R. Powell and J. Aubé, J. Am. Chem. Soc., 2010, 132, 8836 CrossRef CAS.
- M. Szostak, L. Yao and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2078 CrossRef CAS.
- M. Szostak and J. Aubé, J. Am. Chem. Soc., 2009, 131, 13246 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |