DOI:
10.1039/C0OB00342E
(Emerging Area)
Org. Biomol. Chem., 2011,
9, 20-26
Oxidative amide synthesis directly from alcohols with amines
Received
29th June 2010
, Accepted 21st September 2010
First published on 9th November 2010
Abstract
Transition metal catalyzed oxidative amide synthesis directly from primary alcohols and amines is a highly atom economical transformation that evolves hydrogen gas as the only by-product. Several Ru-, Rh-based homogeneous and Ag-based heterogeneous catalysts have been developed for direct amide synthesis. Most of the developed catalysts showed excellent activity with sterically unhindered alcohols and amines; however, limited activity was observed with sterically hindered alcohols or amines, less basic aryl amines, and secondary amines. This account provides an overview of recent advances and challenges in direct amide synthesis.
 Cheng Chen | Cheng Chen was born in Wuhan, P. R. China. He received his BS degree in Applied Chemistry from Wuhan University in 2006. He obtained his MS degree in Analytical Chemistry from the same university in 2008. Presently, he is pursuing his PhD degree in Chemistry under the supervision of Dr Soon Hyeok Hong at Nanyang Technological University in Singapore. His research is focused on ruthenium catalyzed direct amide synthesis from alcohols with amines and development of new organic transformations catalyzed by transition metal complexes. |
 Soon Hyeok Hong | Soon Hyeok Hong received his BS and MS degrees (1999) with Professor Young Keun Chung from Seoul National University. He was then a chemistry full-time instructor at Korea Air Force Academy for his military service (1999–2002). He received his PhD at Caltech with Professor Robert H. Grubbs (2002–2007). After having postdoctoral experience with Professor J. Fraser Stoddart at UCLA and industrial experience at Materia, Inc, he became an assistant professor at Nanyang Technological University in 2008. His research interests include development of efficient, practical, and environmentally friendly synthetic methods and homogeneous catalysts. |
Introduction
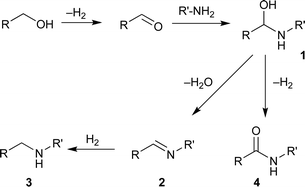
The amide bond plays a key role in organic and biological chemistry. The most common traditional method for the synthesis of the amides is treatment of activated carboxylic acid derivatives with amines.1 Numerous alternative strategies such as the Staudinger reaction,2 the Schmidt reaction,3 and the Beckmann rearrangement4 have been developed. However, in most of these methods, stoichiometric amounts of various reagents are required and equimolar amounts of by-products are produced as waste. Recently, a highly atom economical and environmentally benign method, transition metal catalyzed amide synthesis directly from alcohols and amines, was highlighted.5 The strategy is illustrated in Scheme 1. An alcohol is initially oxidized to the corresponding aldehyde that reacts with an amine to produce a hemiaminal intermediate (1). There are two possible further pathways of the hemiaminal—either it would form an imine (2), which could be subsequently hydrogenated to an amine (3), or would be further oxidized to the corresponding amide (4). The pathway of the dehydration and the hydrogenation of 1, overall alkylation of the amine, has been extensively reported.5–6 Recent examples have shown that further dehydrogenation of 1 producing amides can be achieved instead, depending on the nature of catalysts, ligands, and substrates. It is currently unexplored what properties of the catalytic systems affect an intermediate toward the alkylation or the amidation, therefore more mechanistic investigations will be necessary.5 In this article, recent catalyst development for direct amide synthesis and current challenges in this area are presented.
Oxidative amide synthesis from alcohols and amines is mainly promoted through homogeneous catalysts using Ru- and Rh-based complexes. A heterogeneous Ag-based catalyst was recently reported.
Ru-based catalytic systems
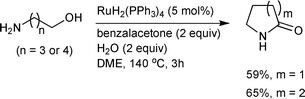
In 1991, Murahashi and Naota reported the first example of the reaction by synthesizing lactams with 1,4- and 1,5-amino alcohols in an intramolecular amidation process (Scheme 2).7 Using RuH2(PPh3)4 as a catalyst and a hydrogen acceptor such as benzalacetone, lactams were formed with good yields. When the reaction was run without the hydrogen acceptor, only cyclic amines were obtained. Two equivalents of water had to be added so as to avoid the formation of cyclic amines if there was a primary amine group in the amino alcohol, while addition of water was not required in the case of amino alcohols bearing a secondary amine group.
 |
| | Scheme 2 | |
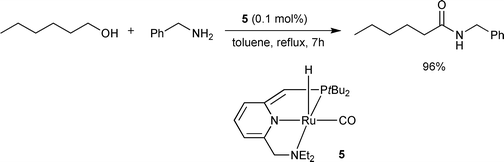
Recently, the Milstein group employed a Ru PNN pincer complex 5 for the direct amide synthesis from alcohols and primary amines without any base, acid promoter, or hydrogen acceptor (Scheme 3).8 It was the first example of allowing the direct amidation of alcohols with amines in an intermolecular fashion. To facilitate the removal of hydrogen gas, the reactions were carried out under a flow of argon. Excellent yields for sterically non-hindered substrates and slightly reduced yields for moderately hindered substrates were obtained. Treatment of primary diamines with alcohols yielded bis-amides in high yields. This catalyst showed a moderate activity for the amidation of aniline, and no activity for the amidation of a secondary amine, dibenzylamine. The essential structure in this catalyst was suggested as being the dearomatized ring which plays an active role in the hydrogen abstraction and liberation process. Complex 5 has been commercialized.
 |
| | Scheme 3 | |
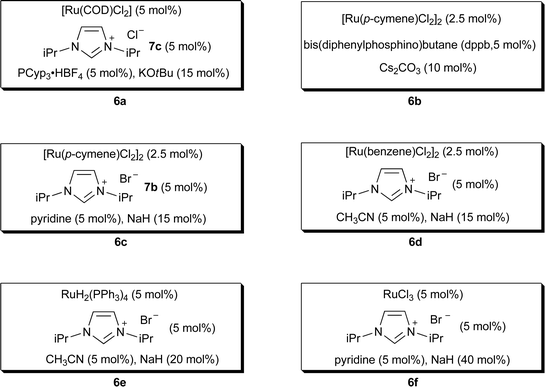
Several in situruthenium catalysts for intermolecular amidation reactions have been developed since Milstein's report (Fig. 1). Madsen and co-workers reported an in situN-heterocyclic carbene (NHC) based ruthenium catalyst.9 This catalytic system (6a), consisting of [Ru(COD)Cl2] (COD = 1, 5-cyclooctadiene), an NHC precursor (1,3-diisopropylimidazolium chloride, 7c), a phosphine ligand such as PCyp3·HBF4 (PCyp3 = tricyclopentylphosphine), and KOtBu, catalyzed reactions efficiently with non-sterically hindered alcohols and primary amines. Notably, optically pure 1-phenylethylamine was converted to the corresponding amide in 70% yield with no observation of racemization (Scheme 4). However, this system exhibited limited activity with aniline and secondary amines.
 |
| | Fig. 1
In situ generated ruthenium catalytic systems. | |
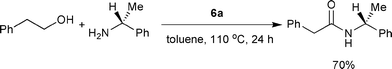 |
| | Scheme 4 | |
It has been logically proposed that direct alcohol amidation goes through aldehydes from the oxidation of alcohols (Scheme 1). However, when they tested the amidation of benzaldehyde with benzylamine, only the corresponding imine was formed without any observation of either the amide or the amine. The imine does not react under the catalytic condition of 6a nor has been detected during the reaction of an alcohol and an amine. Based on the results, it was proposed that these reactions do not go through a free aldehyde but a Ru-bound aldehyde-like species.
Williams and coworkers reported the formation of secondary amides in moderate to good yields using [Ru(p-cymene)Cl2]2, bis(diphenylphosphino)butane (dppb), and Cs2CO3 as the catalytic system (6b) in refluxing t-butanol.10 A hydrogen acceptor such as 3-methyl-2-butanone (2.5 equiv vs. an alcohol substrate) was required to promote the catalysis (Scheme 5). Secondary amines were poor substrates, and in the case of morpholine, a 31% yield of the tertiary amide was obtained with the reaction with 3-phenylpropan-1-ol.
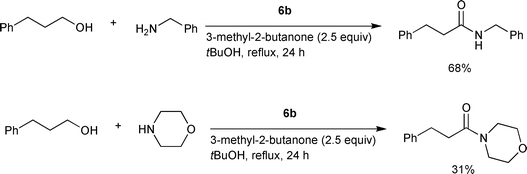 |
| | Scheme 5 | |
Phosphine-free ruthenium catalytic systems (6c and 6d) for effective amide synthesis from alcohols and amines were also reported by Hong and coworkers.11 The catalytic systems consist of [Ru(p-cymene)Cl2]2 or [Ru(benzene)Cl2]2, an NHC precursor (7b), pyridine or acetonitrile, and NaH as a base (6c and 6d). Sterically nonhindered substrates worked smoothly, and moderately hindered ones reacted reasonably well. Limited yields were found for the sterically bulky substrates such as neopentyl alcohol. A few secondary amines were also tested, and good to excellent yields were obtained with less hindered secondary amines such as piperidine, morpholine, and N-benzylmethylamine (Scheme 6). However, no amide was formed from the more sterically hindered dibenzylamine in agreement with Milstein's observations.8 Interestingly, for the reaction between benzaldehyde and benzyl amine with 6c, the corresponding amide (48%) was observed with the concurrent formation of the imine (14%), unlike Madsen's observation that only imine was formed with 6a.9
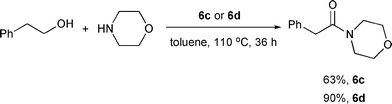 |
| | Scheme 6 | |
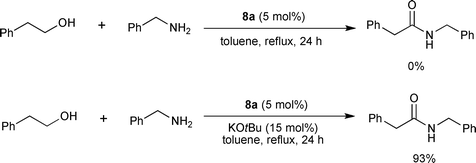
Based on the reported phosphine-free in situ NHC-based Ru catalysts, the Hong group investigated complexes 8a–e (Fig. 2) as precatalysts for the direct amide synthesis.12 When the activity was screened only with the NHC-Ru complexes, there was no formation of the amide from 2-phenylethanol and benzylamine (Scheme 7). It was found that at least 2 equiv of a strong base versus the precatalyst is necessary for the catalytic cycle to proceed. Higher amounts of the base was detrimental to the catalytic activity, suggesting the role of the base is related to the activation of the precatalyst. Among the NHC Ru complexes, 8a and 8c showed better activities, similar to the in situ NHC-based Ru catalysts using 7b or 7c as an NHC precursor.9,11 The amount of base was optimized as 15–20 mol% when 5 mol% of 8a was used for the reaction. Complex 8a showed comparable activity under basic conditions with the previous NHC-based in situcatalytic systems by Madsen9 and Hong.11
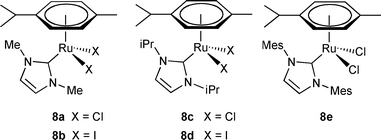 |
| | Fig. 2 Well-defined NHC based ruthenium complexes. | |
 |
| | Scheme 7 | |
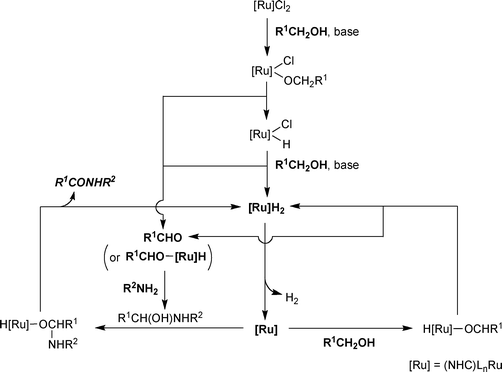
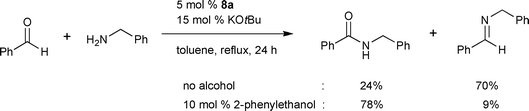
The nature of the active catalyst was investigated with complex 8a under similar catalytic conditions by 1H NMR spectroscopy.12 It was reported that a Ru hydride complex, firstly generated by β-hydrogen elimination of Ru alkoxides formed from the precatalyst and alkoxide, could be an active catalytic intermediate.12 A mechanism involving a Ru(0)/Ru(II) cycle was proposed on the basis of the investigation (Scheme 8), especially elucidating the reason why free aldehyde was not so active under the reported catalytic systems—less efficient formation of active [Ru]H2 from [Ru]Cl2 and an aldehyde without the help of a primary alcohol. When 10 mol% of a primary alcohol was added, the amidation of benzaldehyde with benzylamine proceeded well demonstrating that formation of a catalytically active species with an alcohol is necessary for the amidation of aldehydes (Scheme 9). However, reduced yields from aldehydes and no observation of free aldehyde during the reaction implied that the amide formation from an alcohol might occur through a transition metal bound aldehyde-like species.
 |
| | Scheme 8 | |
 |
| | Scheme 9 | |
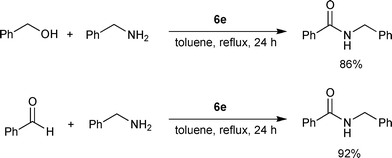
Based on the study of the Ru hydride catalytic intermediates and the Ru chloride precatalyst activation requirement for aldehyde amidation, an in situcatalytic system which can transform either alcohols or aldehydes to amides with amines was developed by the Hong group.13 The catalyst (6e) was generated from readily available RuH2(PPh3)4, an NHC precursor (7b), NaH, and acetonitrile. Both alcohols and aldehydes were efficiently transformed into the amides with 6e (Scheme 10). The previously proposed Ru(0)/Ru(II) cycle was supported by the observation of elimination of hydrogen from a reaction among RuH2(PPh3)4, 7b, and NaH at a reaction temperature of 110 °C. Homogeneous Ru(0) complexes such as Ru3(CO)12 and (η4-1,5-cyclooctadiene)(η6-1,3,5-cyclooctatriene)ruthenium [Ru(cod)(cot)] were also active for the synthesis of amides from alcohols and amines in the presence of 7b and NaH.
 |
| | Scheme 10 | |
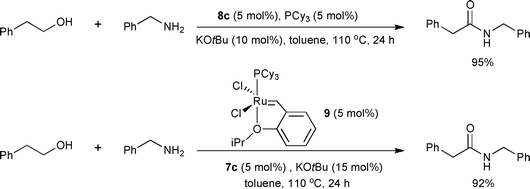
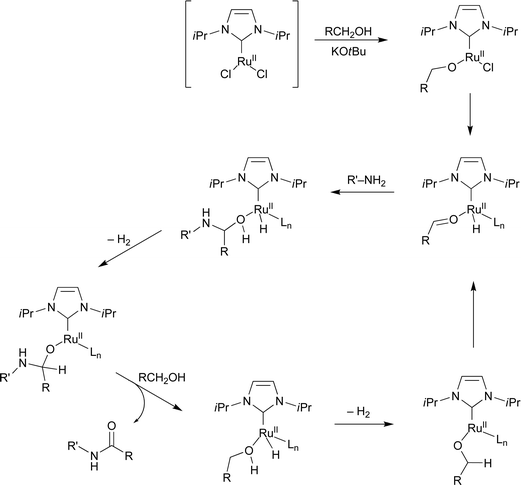
The Madsen group also reported catalytic systems based on the well-defined Ru-NHC complex 8c shortly after the publication of 8a–e as the precatalysts by the Hong group.14 Compared with the reported system,12 they optimized the conditions of 8c with tricyclohexylphosphine (PCy3) as a supporting ligand and a smaller amount of KOtBu (10 mol% vs. 15 mol%, for 5 mol% of 8c, Scheme 11). Moderate to excellent yields for the direct amide synthesis from alcohols and amines were demonstrated. The authors proposed a Ru-bound aldehyde and a Ru-bound hemiaminal as intermediates. It was also pointed out that the oxidation state (+2) of all Ru species remained the same in the whole catalytic cycle (Scheme 12) in contrast to the previous proposition of a Ru(0)/Ru(II) cycle as a possible pathway. The mechanistic question should be investigated further as there is no solid experimental evidence. With the proposition of [(IiPr)RuII] as the catalytically active components, Ru olefin metathesis catalysts were screened for the direct amidation of primary alcohols. The best results were obtained with Hoveyda–Grubbs 1st-generation metathesis catalyst 9 with 7c and KOtBu. The three catalyst systems based on 6a, 8c, and 9, do not show significant differences in reactivity, supporting that the same catalytically active species, [(IiPr)RuII], is operating under the NHC-based Ru catalyst systems.
 |
| | Scheme 11 | |
 |
| | Scheme 12 | |
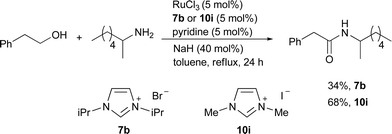
With the similar hypothesis of [(NHC)Ru] as a catalytically active species, another in situcatalyst system (6f) using the more economical RuCl3, an NHC precursor, and pyridine in the presence of NaH was reported.15 Moderately hindered substrates are more reactive when the less bulky NHC precursor 10i was used instead of 7b (Scheme 13). The optimal condition was achieved with 40 mol% NaH and it was claimed that NaH in this system might not only be used to generate NHCs from the corresponding imidazolium salts but also as a reducing agent that reduces Ru(III) to Ru(II) or Ru(0). The authors also found NHC-based olefin metathesis catalysts active for the alcohol amidation with amines, which also demonstrated that the oxidative amide formation can be catalyzed by a Ru complex with an NHC ligand under basic conditions.
 |
| | Scheme 13 | |
 |
| | Scheme 14 | |
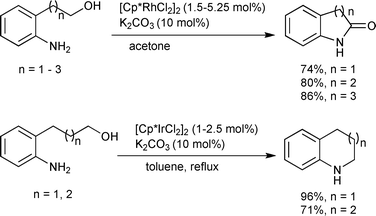
A Rh-based catalyst 11 for intermolecular amide synthesis was developed by Grützmacher and co-workers (Scheme 15).18 A hydrogen acceptor such as methylmethacrylate (MMA) was required to generate primary and secondary amides in excellent yields. The reaction occurs under much milder conditions than with the Ru-based catalyst systems, even at room temperature. This method has good functional group tolerance and chemoselectivity with low catalyst loadings. The amido function in 11 is the Lewis basic site which may be crucial for the catalytic cycle. A computational study illustrated that the rhodium monohydride species is an important intermediate in the whole cycle. Notably, this was the first example of the synthesis of primary amides directly from primary alcohols and ammonia. The Williams group previously reported the primary amide synthesis from alcohols with hydroxylamine in one pot as two step processes, based on the oxidation of an alcohol followed by the Beckmann rearrangement of the oxime intermediate generated from the aldehyde and hydroxylamine, which is catalyzed by [Cp*IrCl2]2 and Cs2CO3 (Scheme 16).19
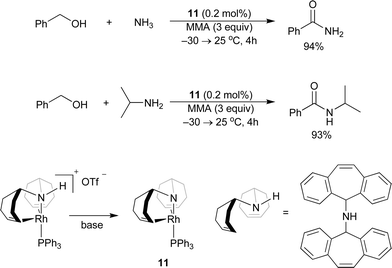 |
| | Scheme 15 | |
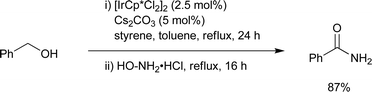 |
| | Scheme 16 | |
Ag-based heterogeneous catalyst
A heterogeneous Ag-based catalyst for the direct amidation of alcohols with amines was reported by Shimizu and coworkers (Scheme 17).20 The alumina-supported silver cluster with Cs2CO3 was active for the amide synthesis with primary amines, cyclic secondary amines, and less sterically hindered non-cyclic secondary amines such as N-benzylmethylamine. Mechanistic investigations suggested that the reaction goes through an aldehyde-like species that was adsorbed on the catalyst, as there was no amide formation from an ester, a free aldehyde, or imine. Subsequent attack on the aldehyde-like species by an amine afforded the hemiaminal, and then the amide was formed after hydride elimination by the silver cluster (Scheme 18). Based on these kinetic studies, C–H cleavage of the alkoxide or hemiaminal by the silver cluster was proposed as the rate-determining step. A cooperation of coordinatively unsaturated silver site and acid–base site of Al2O3 support was suggested from the studies on the structure–activity relationship.
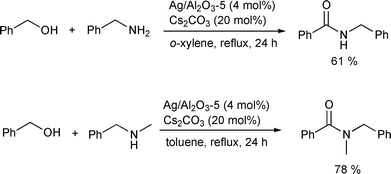 |
| | Scheme 17 | |
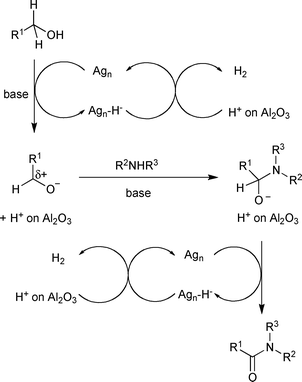 |
| | Scheme 18 | |
Conclusion and future challenges
In recent years, Ru- and Rh- based homogeneous and Ag-based heterogeneous catalysts have been developed for direct amide synthesis from alcohols and amines. Although it is an atom economical transformation producing hydrogen as the sole by-product, there are many challenges in this area to widely apply the emerged methodology to the important amide bond formation in organic synthesis instead of the traditional amide syntheses. Most of the developed catalysts showed excellent activity with sterically unhindered alcohols and amines; however, limited activity was observed with sterically hindered alcohols or amines, less basic aryl amines, and secondary amines. Also, the necessity of special handling of expensive metal complexes and ligands in many cases need to be overcome from the environmental and economic point of view. Mechanistic investigation to understand the exact nature of the catalysts is highly desirable in order to devise more active and practical catalytic systems.
References
-
(a) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS
 ;
(b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS ;
(c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC .
;
(b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS ;
(c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC .
-
(a) M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef ;
(b) F. Damkaci and P. DeShong, J. Am. Chem. Soc., 2003, 125, 4408–4409 CrossRef CAS ;
(c) Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353–1406 CrossRef CAS .
-
(a) T. Ribelin, C. E. Katz, D. G. English, S. Smith, A. K. Manukyan, V. W. Day, B. Neuenswander, J. L. Poutsma and J. Aubé, Angew. Chem., Int. Ed., 2008, 47, 6233–6235 CrossRef CAS ;
(b) S. Lang and J. A. Murphy, Chem. Soc. Rev., 2006, 35, 146–156 RSC ;
(c) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS ;
(d) J. Aubé and G. L. Milligan, J. Am. Chem. Soc., 1991, 113, 8965–8966 CrossRef CAS .
-
(a) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599–3601 CrossRef CAS ;
(b) M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894–2897 CrossRef CAS .
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS .
-
(a) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS ;
(b) M. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS ;
(c) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC ;
(d) A. Tillack, D. Hollmann, D. Michalik and M. Beller, Tetrahedron Lett., 2006, 47, 8881–8885 CrossRef CAS ;
(e) K. Fujita, Y. Enoki and R. Yamaguchi, Tetrahedron, 2008, 64, 1943–1954 CrossRef CAS ;
(f) S.-I. Murahashi, K. Kondo and T. Hakata, Tetrahedron Lett., 1982, 23, 229–232 CrossRef CAS ;
(g) R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC ;
(h) Y. Watanabe, Y. Tsuji, H. Ige, Y. Ohsugi and T. Ohta, J. Org. Chem., 1984, 49, 3359–3363 CrossRef CAS ;
(i) Y. Watanabe, Y. Morisaki, T. Kondo and T. Mitsudo, J. Org. Chem., 1996, 61, 4214–4218 CrossRef CAS ;
(j) M. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS and more references therein.
- T. Naota and S.-I. Murahashi, Synlett, 1991, 693–694 CAS .
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS .
- L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef CAS .
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667–2670 CrossRef CAS .
- S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS .
- Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374–1378 CrossRef CAS .
- S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002–3006 CrossRef CAS .
- J. H. Dam, G. Osztrovszky, L. U. Nordstrøm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820–6827 CrossRef CAS .
- S. C. Ghosh and S. H. Hong, Eur. J. Org. Chem., 2010, 4266–4270 CrossRef CAS .
- K. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785–2788 CrossRef CAS .
- K. Fujita, K. Yamamoto and R. Yamaguchi, Org. Lett., 2002, 4, 2691–2694 CrossRef CAS .
- T. Zweifel, J. V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS .
- N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73–75 CrossRef CAS .
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.