Computational and experimental investigations of mono-septanoside binding by Concanavalin A: correlation of ligand stereochemistry to enthalpies of binding†
Michael R.
Duff Jr.
,
W. Sean
Fyvie
,
Shankar D.
Markad
,
Alexandra E.
Frankel
,
Challa V.
Kumar
*,
José A.
Gascón
* and
Mark W.
Peczuh
*
Department of Chemistry, The University of Connecticut, 55 North Eagleville Road, Storrs, CT 06269, USA. E-mail: mark.peczuh@uconn.edu
First published on 18th November 2010
Abstract
Structure-energy relationships for a small group of pyranose and septanose mono-saccharide ligands are developed for binding to Concanavalin A (ConA). The affinity of ConA for methyl “manno” β-septanoside 7 was found to be higher than any of the previously reported mono-septanoside ligands. Isothermal titration calorimetry (ITC) in conjunction with docking simulations and quantum mechanics/molecular mechanics (QM/MM) modeling established the specific role of binding enthalpy in the structure–energy relations of ConA bound to natural mono-saccharides and unnatural mono-septanosides. An important aspect in the differential binding among ligands is the deformation energy required to reorganize internal hydroxyl groups upon binding of the ligand to ConA.
Introduction
The integral role that protein–carbohydrate interactions play in a variety of biological processes is now well established.1–5 Examples include protein-folding, viral attachment to host cells, immunomodulation, enzymes involved in carbohydrate metabolism, and cell attachment to the extra-cellular matrix, among others. Because of the potential benefits, inhibiting certain protein–carbohydrate interactions that lead to cellular pathologies can provide a valuable entry into new therapeutics. A soluble polysaccharide ligand of galectin-3 (gal-3), for instance, was recently approved for clinical trials to treat multiple cancers.6,7 The release of new influenza viruses from a host cell involves breaking up protein–carbohydrate interactions viahydrolysis of the polysaccharide. Oseltamivir (Tamiflu®) is a >$1b/year carbohydrate analog8 that works by inhibiting the key glycosidase in the process, a neuraminidase.9 The relevance of protein–carbohydrate interactions in biology, therefore, motivates fundamental investigations on the parameters that govern their association and also on the development of new carbohydrate analogs as potential competing ligands.A number of weak non-covalent forces, working in concert, underpin the binding of carbohydrates by lectins and glyco-enzymes.10–12Hydrogen bonding, van der Waals interactions, and desolvation of the ligand and binding site residues have all been shown to contribute to protein–carbohydrate associations.13–16 These forces bear a connection to the dual nature of carbohydrates themselves. The hydroxyl groups on a given sugar residue are critical for H-bonding with its protein partner while also being important for solvation. Hydroxyl groups form a H-bonding “ribbon” around the perimeter of the monosaccharide residue. Alternatively, the C–H bonds collectively define apolar patches that can associate with the π-system of aromatic amino acid residues. Solvation and desolvation of apolar patches is distinct from solvation of the hydrogen bonding array. Specific arrangements of H-bonding arrays and apolar patches as defined by specific sugar residues alone or as part of an oligosaccharide then define how a protein binds to a carbohydrate.
Synthetic analogs of pyranose sugars (e.g., oseltamivir) have been extensively studied. In general, such analogs should find use in glycobiology and as therapeutic leads.17 The literature contains only a few reports, however, of septanoses being used as analogs of natural pyranose sugars to investigate protein–carbohydrate interactions.18–20 Tauss et al.20 reported on the ability of p-nitrophenylL-idoseptanosides 1 and 2 to serve as substrates of exo-glycosidases. Both α- and β- anomers of the septanoside were prepared for the investigation. The L-idoseptanosides (1 and 2) were chosen because they share the same stereochemistry as D-glucose from C1–C4 (Fig. 1). Molecular modeling studies showed that minimized structures of 1 and 2 positioned their aglycones and C2–C4 hydroxyl groups in similar orientations relative to 3 and 4, respectively.20 As measured by the overall efficiency of the enzymatic reaction using α-glucosidase, 1 was a poor substrate (kcat/KM = 24 s−1 M−1) when compared to the pyranose substrate 3 (kcat/KM = 2.4 × 104 s−1 M−1). The data also showed a factor of 10 difference in the relative binding affinities of 1versus3 (KM = 2.9 and 0.27 mM, respectively). Similar results were obtained when comparing the β-glycosides 2 and 4 using β-glucosidase. Albeit with low affinity, even in comparison to low affinities of the “native” substrates, the results of this investigation showed that natural glycosidases could, in fact, bind septanosides and achieve enzymatic turn-over in the hydrolysis of these substrates.
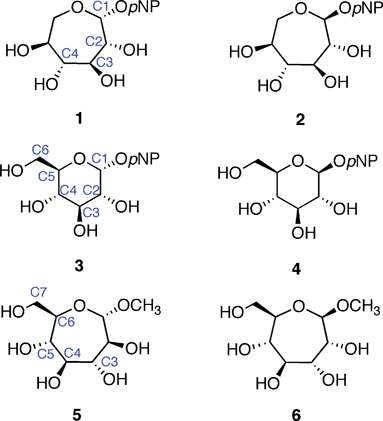 | ||
| Fig. 1 Septanose and pyranose glycosides: L-idoseptanosides (1, 2), D-glucosides (3, 4), D-glycero-D-septanosides (5, 6). | ||
The binding of methyl β-septanosides such as 6 to the plant lectin Concanavalin A was previously reported by this laboratory. Structural information from X-ray crystallographic investigations of Concanavalin A (ConA) complexed with α-pyranose monosaccharides21–26 showed that the primary contacts between the ligand and the protein were along C3–C6. Our design of septanose ligands therefore included an exocyclic hydroxymethyl group (e.g., C7 in 5 and 6). Association constants obtained by Isothermal Titration Calorimetry (ITC) showed that methyl β-septanoside 6 was a weak ligand of ConA (Ka = 450 M−1). For comparison, methyl α-glucoside 9 and methyl α-mannoside 10 (their six-member ring counterparts; Fig. 2) gave affinity constants of 2000 and 7900 M−1, respectively. 1H NMR Saturation Transfer Difference (STD) competition experiments confirmed that binding to 6 was at the pyranoside binding site. The investigation showed that β-septanosides were bound by ConA but α-septanosides such as 5 were not. Remarkably, this selectivity for anomeric configuration is opposite to that observed for ConA binding to pyranosides. That is, α-pyranosides are ligands of ConA while β-pyranosides are not.19 An important conclusion that was drawn from these preliminary studies of protein–septanose interactions was that, based on a given protein, septanoses may be bound with low affinity. In both the glycosidase and the lectin examples, presentation of specific hydroxyl groups by the septanosides to mimic the “target” pyranosides was shown to be important for binding. It remained unclear exactly how the changes to individual stereocenters of the ligands correlated with their ability to be bound by ConA.
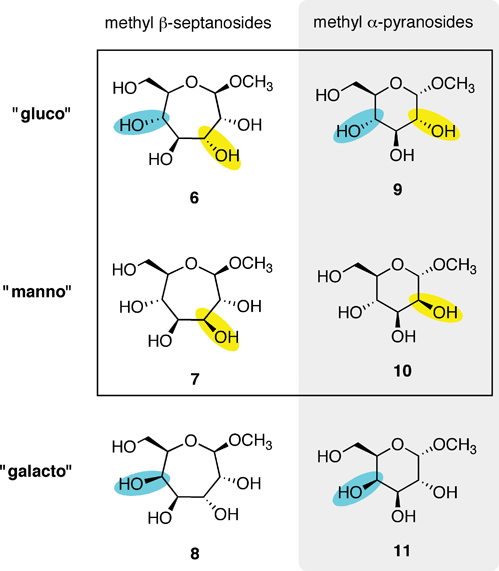 | ||
| Fig. 2 Methyl β-septanosides 6–8 and methyl α-pyranosides 9–11 used as ligands of ConA. Each row is organized by related ring stereochemistries: “gluco”, “manno” and “galacto”. Changes in stereochemistry are denoted by yellow/blue highlights. | ||
We endeavored, therefore, to understand how septanosides interacted with ConA more precisely and how this association corresponded to the way pyranosides are bound. In the same way that the initial mono-septanoside ligand of ConA could loosely be considered a ring expanded analog of D-glucose (Fig. 1), we expected ring expanded analogs of D-mannose and D-galactose to behave like their pyranoside counterparts. Reported here is the calorimetric determination of association constants for ConA binding to several new seven membered ring sugar analogs. We further show the origin of the relevant protein–ligand interactions as they pertain to enthalpic contributions in detail. This is accomplished by correlating the binding data with structural information from docking and Quantum Mechanical/Molecular Mechanical (QM/MM) calculations. We show that there is a very good qualitative correlation between the gas-phase QM/MM binding energy and the experimental enthalpy of binding. This allows us to make a structure–energy correlation. Further, the implications of the results in terms of utilizing septanoses as analogs of pyranoses are discussed.
Methyl glycoside ligands
Methyl glycosides used in this investigation are shown in Fig. 2. The absolute affinities of ConA for monosaccharides are generally low; however, the literature has several examples of approaches for increasing this affinity via polyvalent presentation of the ligand.27–31 Moreover, ConA also has a higher affinity for the Man α-1-3,1-6 trisaccharide, which is the biologically relevant ligand.32–34 We chose to analyze binding between a series of monosaccharides because access to the septanoside ligands was straightforward and structure–energy relationships could be developed. Ring expanded analogs 6–8 were chosen for comparison to each other and also to their pyranose counterparts 9–11. Fig. 2 organizes the ligands based on whether the ligand is a septanoside or pyranoside (columns) and also on ring stereochemistry (rows). Each series reflects the stereochemical configuration of C2–C5 (pyranose numbering) of glucose, mannose, and galactose, respectively.Methyl β-septanoside 6 of the gluco- series serves as a starting point for comparison of the monosaccharides in this study. As previously mentioned, 6 was the ring-expanded ligand with the highest affinity for ConA in our earlier investigation. The new methyl β-septanosides 7 and 8 vary one stereocenter on the ring relative to 6; in manno- analog 7, C3 has been inverted (septanose numbering) whereas in galacto- analog 8 C5 was inverted. Stereocenters C3–C6 in 6–8 correspond to the C2–C5 stereocenters of 9–11. Synthesis of methyl β-septanosides 6–8 was accomplished by the epoxidation and methanolysis of carbohydrate based oxepines35 followed by removal of the protecting groups on the C3–C7 hydroxyls.36,37
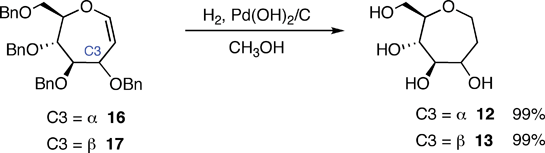
Additional molecules were prepared in an effort to obtain further details regarding the structure–energy relationships that had been observed (Fig. 3). The first pair, oxepanes 12 and 13, were prepared by hydrogenation of their oxepine precursors along with concomitant hydrogenolysis of the benzyl ether protecting groups (Scheme 1). Methyl α-septanoside 14 and methyl 2-O-methyl β-septanoside 15 were also targeted. Preparation of 14 from a mannose-derived oxepine had been described earlier.37Methyl 2-O-methyl septanoside 15 was prepared by epoxidation and methanolysis of the glucose-derived oxepine followed by methylation of the C2 hydroxyl group and subsequent debenzylation (Scheme 2) (The Experimental Section details the synthesis of newly reported compounds 12, 13, and 15.). We had originally planned to synthesize a family of C2-acylated septanosides by this route where acylation at C2 would have substituted for the alkylation step. However, acyl migration proved to be a complicating side-reaction during the hydrogenolysis of the benzyl protecting groups and so attempts at preparing 2-O-acyl analogs of 6 were abandoned.
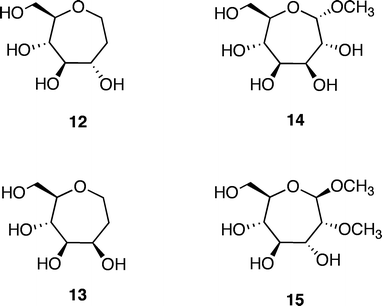 | ||
| Fig. 3 Additional ligands of ConA used in the investigation: Oxepanes 12 and 13 derived from D-glucose and D-mannose, respectively; methyl “manno” α-septanoside 14 and methyl 2-O-methyl “gluco” β-septanoside 15. | ||
Computational approach
We turned to a computational approach to gain greater insight into the atomic level interactions between ConA and the monosaccharide ligands that were studied by ITC. We were especially interested in discerning the differences in affinity between the septanoside ligands and their pyranoside counterparts. Modeling of all protein–ligand complexes was based on the X-ray structure of Concanavalin A (ConA) with 9 (PDB access code: 1GIC chain A).23 We analyzed the binding of seven ligands (5–10, 15), for which thermodynamic data had been obtained (Table 1). These complexes span the entire range of binding affinities observed, and can help understand the origin of relevant interactions. To take advantage of the steric constraints imposed by the protein cavity we used the co-crystal (ConA bound to 9) to create the different stereoisomers by modifying the existing ligand in situ. After in situ modification with the program Maestro38 and removal of all crystallographic waters except Wat335 (see discussion below), each ligand (including 9) was re-docked with the program Glide,39 always maintaining the structure of the protein frozen. The docking algorithm in Glide allows for flexibility and bond rotation and uses scoring functions predominantly based on a molecular mechanics (MM) force field.| Ligand | K a/M−1 | N | ΔG/kcal mol−1 | ΔH/kcal mol−1 | TΔS/kcal mol−1 |
|---|---|---|---|---|---|
a
Titrations were run at 298 K. [ConA] was between 0.200–0.300 mM and [5–15] was between 15–30 mM. ConA was dimeric under the experimental conditions. Buffer: 50 mM 3,3-dimethylglutarate pH 5.2, 250 mM NaCl, 1 mM CaCl2, 1 mM MnCl2.
b Data from ref. 19
c Data fit with the ConA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand stoichiometry manually fixed at 1:1; therefore N = 1.
d Data from ref. 46. NB = no binding was detected under these conditions. :ligand stoichiometry manually fixed at 1:1; therefore N = 1.
d Data from ref. 46. NB = no binding was detected under these conditions.
|
|||||
| 5 b | NB | — | — | — | — |
| 6 b | 5.2 ± 0.3 × 102 | 3.2 ± 0.9 | −3.7 ± 0.1 | −0.83 ± 0.1 | 2.9 ± 0.2 |
| 6 b | 4.5 ± 0.7 × 102 | 1c | −3.6 ± 0.09 | −2.7 ± 0.2 | 0.95 ± 0.3 |
| 7 | 8.4 ± 0.4 × 102 | 0.9 ± 0.1 | −4.0 ± 0.03 | −5.5 ± 0.6 | −1.5 ± 0.6 |
| 8 | NB | — | — | — | — |
| 9 b | 2.0 ± 0.2 × 103 | 0.87 ± 0.03 | −4.5 ± 0.03 | −3.5 ± 0.9 | 0.98 ± 0.2 |
| 10 b | 7.9 ± 1.0 × 103 | 0.83 ± 0.18 | −5.5 ± 0.1 | −4.5 ± 0.03 | 1.1 ± 0.2 |
| 11 d | NB | — | — | — | — |
| 14 | NB | — | — | — | — |
| 15 | 4.3 ± 1.0 × 102 | 9.1 ± 3.1 | −3.6 ± 0.1 | −0.13 ± 0.05 | 3.4 ± 0.2 |
| 15 | 1.9 ± 0.3 × 102 | 1c | −3.1 ± 0.1 | −1.4 ± 0.2 | 1.7 ± 0.3 |
Docking computations were followed by QM/MM energy minimizations using Qsite,40 where the ligand and Asp208 were treated at the quantum mechanical (QM) level and the rest of the protein at the MM level. Cuts between the QM and MM region were treated with the frozen-orbital method as implemented in Qsite. The MM region was treated with the OPLSAA force field,41 and the QM region with Density Functional Theory (DFT). The functional B3LYP and basis set 6-31g* were used in all QM calculations.
We considered two sets of calculations, one in which the protein remained frozen in its crystallographic position while only the ligand was relaxed, and another where the ligand and the specific residues Asn14, Leu99, Tyr100, Asp208, and Arg228 were relaxed. It will be shown below that both sets of calculations produce similar results and both lead to the same analysis regarding the differential binding among the ligands.
The gas phase binding energy (Eb) is defined as:
| Eb = EQM/MM − E°QM/MM − EQM , | (1) |
| ΔH = ΔH0 + (ΔHcomplex − ΔHlig − ΔHprot). | (2) |
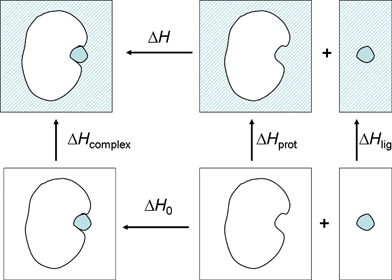 | ||
| Fig. 4 Thermodynamic cycle describing the separation of the observed binding enthalpy in solution into the gas phase binding enthalpy and solvent reorganization enthalpy. | ||
In the present work, we have focused on the relative enthalpy of binding, under the assumption of a rigid cavity (ΔΔHprot = 0):
| ΔΔH = ΔΔH0 + (ΔΔHcomplex − ΔΔHlig). | (3) |
We now argue that, due to the similar nature of the ligands considered, ΔΔHcomplex − ΔΔHligand is negligible. This would not be the case for structurally dissimilar ligands. Our assumption is supported by studies carried out by Toone's group on protein–carbohydrate binding.43 In that work, a direct measure of the solvent reorganization contribution to enthalpy was obtained by measuring the binding enthalpies in D2O relative to H2O. Key to our study was that the difference in the enthalpy of solvation (ΔΔHlig) across several monosaccharides (including methyl pyranosides 9 and 10, for example) was of the order of 0.2 kcal mol−1.44,45 The other contributor to the solvent reorganization is the solvation of the protein–ligand complex (ΔHcomplex). Although no experimental results for this contribution to the protein–ligand complexes have been determined, it is logical to assume that, since all ligands utilize a similar set of interactions when binding to the protein (see results below), ΔΔHcomplex should be of a similar magnitude as ΔΔHlig but with the opposite sign. This assumption is also supported a posteriori by the good qualitative and quantitative correlation between the gas-phase QM/MM calculations and the experimental enthalpies of binding (see Table 2). Although inclusion of solvent effects, via implicit or explicit solvent treatments would be certainly desirable and would likely yield better results, the computational cost of such a treatment would be significantly higher at the present level of theory (i.e. DFT-QM/MM). Thus, assuming a constant effect of the solvent throughout the various ligands as argued above, the absolute binding energies were no longer meaningful. Instead, our interest was focused on relative binding energies (ΔEb), rather than absolute binding energies, which we assume, as argued above, to be a good approximation of ΔΔH0. Again, we emphasize that for general ligands this may not be the case, but because of the incremental difference between any two monosaccharides, this approximation appears well founded.
| Ligand | E d /kcal mol−1 | E rb /kcal mol−1 | E b /kcal mol−1 | ΔEb/kcal mol−1 | ΔΔHa (exp.)/kcal mol−1 |
|---|---|---|---|---|---|
| a ΔΔH = ΔH(ConA·ligand) − ΔH(ConA·7). | |||||
| 5 | 23.1 | −87.1 | −64.0 (−58.3) | 8.8 (9.0) | no binding |
| 6 | 18.6 | −86.2 | −67.6 (−61.8) | 5.2 (5.5) | 4.67/2.8 |
| 7 | 14.4 | −87.2 | −72.8 (−67.3) | 0.0 (0.0) | 0.0 |
| 8 | 24.3 | −80.2 | −55.9 (−51.9) | 16.9 (15.4) | no binding |
| 9 | 13.7 | −84.8 | −71.1 (−66.2) | 1.7 (1.1) | 2.0 |
| 10 | 14.3 | −86.1 | −71.8 (−66.3) | 1.0 (1.0) | 1.0 |
| 15 | 17.1 | −85.6 | −68.5 (−62.6) | 4.3 (4.7) | 5.37/4.1 |
It is also important to mention recent work from the Woods laboratory on the involvement of structural waters in the binding of ConA to the natural mannose trisaccharide and a related analog.15 That work addressed the role of a conserved boundwater molecule which makes hydrogen bonds to Asn14, Arg228, and Asp16 and remains bound upon ligand binding. This water becomes relevant as the central mannosyl residue of the trisaccharide possesses a hydroxyl group very close to that water. It was shown that the replacement of this group by a hydroxyethyl side chain resulted in a more strained conformation of this water molecule with respect to its cavity affecting the enthalpy of binding. All other waters in the binding site appeared to be displaced upon binding. Therefore, considering this important observation we included this water molecule (Wat335 in 1GIC.pdb) in all QM/MM calculations. Hydrogen atoms were added to this molecule and their orientation optimized to form hydrogen bonds to Asn14, Arg228, and Asp16 following the findings of Kadirvelraj et al.15
Results and discussion
ITC Studies
The binding interactions between ConA and specific monosaccharides have been quantified by calorimetric titrations where the heat produced or absorbed during the titration has been detected. The ITC data for one of the substrates (7) are shown in Fig. 5. Each addition of the ligand solution to the solution of ConA resulted in the release of heat, and a concomitant peak is observed in the data trace (Fig. 5, left). As the titration progressed, the heat released diminished progressively due to the gradual saturation of the available binding sites. The area data are then integrated, analyzed, and binding isotherms constructed (Fig. 5, right).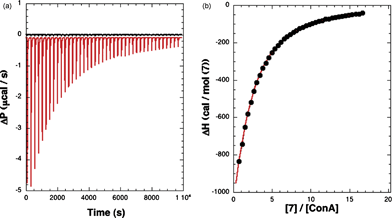 | ||
| Fig. 5 ITC of methyl “manno” β-septanoside 7 into ConA. Titration conditions are given in Table 1. (Left) Raw injection traces from titration of 7 into ConA (red) and dilution of 7 into buffer (black). (Right) Titration curve generated from raw data. | ||
Analysis of the data with specific binding models that gave best fits to the data were used to obtain the binding enthalpy (ΔH), binding entropy (ΔS), binding free energy (ΔG), affinity constant (Ka), and the number of binding sites per ConA (N) extracted (Table 1). Such analysis of the ITC data with other substrates has been carried out and resulting thermodynamic parameters collected in Table 1, which are discussed below.
Binding of the pyranoside ligands 9–10 and septanosides 6, 7, and 15 to ConA was exothermic. A notable parallel between the trend in binding affinities for methyl β-septanosides 6–8 and methyl α-pyranosides 9–11 became apparent. The rank ordering of ConA affinities for the pyranosides is methyl α-mannoside 10 (7900 M−1) followed by methyl α-glucoside 9 (2000 M−1). Methyl α-galactoside 11 is not bound by ConA.46 The same ordering of affinities for methyl β-septanosides held true. Manno- analog 7 (850 M−1) was bound more tightly than gluco- analog 6 (520 M−1) and the galacto- analog 8 showed no binding in our assay. The data clearly suggested that the methyl β-septanosides bind in a manner analogous to methyl α-pyranosides based on this parallel. A key point is that the septanoside binders were consistently poorer ligands (by factors of 5–10) compared to the pyranoside binders. One objective of the computational work presented here was to develop a way to rationalize this observation.
To our surprise, 12 and 13 were very poor ligands for ConA. Weak exothermic binding was observed in the corrected ITC traces, but curve fitting of the data gave poor fits and highly variable association constants and stoichiometries. We interpreted this to mean that 12 and 13 were binding, but with Ka values that were significantly lower than the ones reported for the other ligands in Table 1. The oxepanes were intended to measure if the aglycone moiety contributed to binding. Preliminary manual overlays of 5 and 6 in the binding pocket of the 9–ConA co-complex19 suggested that the methyl aglycone of 5 was making unfavorable van der Waals contacts with the protein that were likely responsible for the lack of binding between ConA and 5. Such an unfavorable interaction was largely, but not completely, eliminated in 6. We reasoned that removing the aglycone entirely (and also the C2 stereocenter) could maintain the important contacts with ConA at C3–C7 while removing the unfavorable aglycone–protein interaction. We also anticipated that the absence of an anomeric center could influence the conformational behavior of the ring. Therefore, comparison of the binding data of 12 and 13 with those of 6 and 7 suggested that the anomeric centers of 6 and 7 were, in fact, crucial for binding to ConA.
Titration of ConA with methyl “manno” α-septanoside 14 was to confirm that, even in the higher affinity series (i.e.methyl “manno” β-septanoside 7), stereochemistry at the anomeric center was in fact critical for binding. Lack of any discernable binding of 14 supported this assumption. Methyl β-2-O-methyl-septanoside 15 was used to gauge whether addition of specific functional groups at the C2 position could increase affinity for ConA. A preliminary manual overlay of 15 at the binding site on ConA suggested that methyl β-septanoside 6 was “de-phased” relative to its pyranose counterpart 9;19 that is, C2 of the septanose partially overlapped with the anomeric carbon (C1) of 9 in the binding pocket. Acylation/alkylation at C2 of the septanose was therefore intended to mimic the aglycone of the pyranose. Binding by ConA to 15 was of a similar magnitude to that observed for the related methyl “gluco” β-septanoside 6; it seemed that functionalization at the C2 hydroxyl group increased the affinity for ConA only marginally. Based on the modest binding behavior, the complications with acylation at C2 and anticipated difficulties of alkylation with other groups at this position, more detailed structure affinity relationships at this position were not pursued. Overall, the experimental binding data provided a model for the interaction where affinity depended on individual stereocenters of the monosaccharide (both pyranose and septanose) ligands and also the presence of an anomeric center, presumably as a rigidifying feature.
It should be noted that the concentration of ConA used in the above titrations, when combined with low affinities of ligands 6 and 15, resulted in c parameters of 0.10 to 0.16.47 These values are outside the optimal experimental window (c = 10–500). A consequence of the low c values is that the binding stoichiometry, expressed as parameter N in Table 1, deviated from the expected value of 1.48 As was done previously, the data for 15 were fit where the N value was manually set at 1. This was done to approximate the ΔH value in such a scenario. The magnitude of ΔH can be underestimated when the c value is low. As presented below, the experimental ΔH values where N was either allowed to float or was fixed were correlated to the calculated ΔH values separately.
Binding energy contributions
In order to analyze the origin of the different binding energies amongst the ligands, we separated the binding energy into two contributions. First, we determined a deformation energy (Ed), defined as the energy required to deform the gas-phased optimized structure into the conformation adopted by the ligand inside the protein. Second, we computed the rigid-binding energy (Erb), using the deformed ligand for the gas phase calculations in eqn (1), instead of the relaxed one. Thus, the binding energy can also be expressed as:| Eb = Ed + Erb. | (4) |
Table 2 shows the computed relative binding energies for ConA bound to 5–10 and 15 (ΔEb) along with their decomposition in the various terms. Since we were particularly interested in the deformation effect of the ligand alone, we made the separation of rigid binding and deformation using the computational model which assumes a frozen protein cavity. Thus, for the set of calculations with a relaxed cavity, we only report the relative binding energies. The reference system was chosen to be the complex between ConA and methyl “manno” β-septanoside 7 because it had the greatest enthalpic contribution to binding of the ligands investigated (Table 2). The binding energies show a very good qualitative agreement with the experimental enthalpy values (ΔΔH) for all ligands and for both sets of calculation (frozen protein and relaxed cavity). In both experiment and computation, the complex of ConA with 7 displayed the largest enthalpy of binding. The computed values of +17 kcal mol−1 for ConA·8 and +9 kcal mol−1 for ConA·5 were also consistent with the lack of binding observed by ITC (Table 1). Complexes that showed intermediate ΔEb and ΔΔH values for ConA bound to 6, 9, 10, and 15 also correlated well (Fig. 6). The two plots in Fig. 6 demonstrate the correlation. The solid squares correspond to the case with a frozen cavity while the open triangle corresponds to the case with a relaxed cavity. The experimental ΔH values used in this plot are those in which N was allowed to float. These two datasets gave correlation coefficients of 0.968 and 0.965, respectively. Both coefficients are of sufficient magnitude to indicate that both models captured the most important aspects of the interaction. If the same correlation is carried out with the ΔΔH values for ligands 6 and 15 where N was fixed at 1, a correlation of 0.885 and 0.865 is obtained, respectively, suggesting that fixing the N values may, in fact, overestimate the experimental ΔH values.
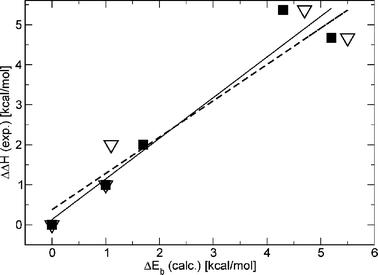 | ||
| Fig. 6 Correlation between the computed binding energy and the experimental enthalpy of binding. Solid line represents a linear regression (correlation coeff. = 0.968) using the solid square data. Dashed line represents a linear regression (correlation coeff. = 0.962, see text for discussion) for open triangles data. | ||
Structure–energy relationships
The correlation between the computational binding energies and the experimentally determined ΔΔH values gave us confidence to use the computed structures to piece together the protein–ligand interactions contributing to the range of binding energies observed. Fig. 7 shows the Glide-QM/MM structure of ConA bound to methyl α-glucoside 9 according to the frozen cavity QM/MM model. The calculated complex reproduces the co-crystal structure almost exactly, resulting in a root mean square deviation (RMSD) of 0.3 Å. One immediate observation is that Asp208 is the acceptor of two hydrogen-bonds from the glycoside, a feature common to all the ligands considered. In this regard, it should be expected that elimination of Asp208 would cause a substantial decrease in binding affinity.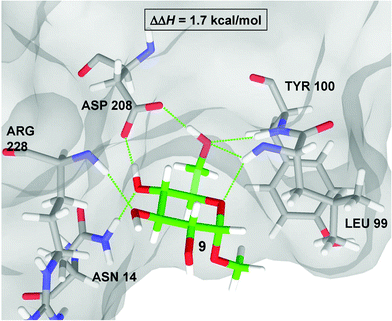 | ||
| Fig. 7 QM/MM structure of the ConA·9 complex. All immediate residues form hydrogen-bonds with the methyl α-D-glucoside are displayed as thin green lines. | ||
The enthalpic contribution to the binding affinity in this complex primarily arises from the seven hydrogen-bonds which connect it to a number of residues in the ConA binding pocket (see Fig. 7). In particular, 9 makes two hydrogen bonds to the side chain of Asp208, two to the backbone of Leu99, one to each of the backbones of Arg228 and Tyr100, and one to the side chain of Asn14. The side chain of Leu99 is positioned so that α-pyranosides such as 9 matched the binding pocket, and escape unfavorable van der Waals contacts between the aglycone and Leu99. The aglycone of a β-pyranosides, on the other hand, would need to occupy the same space as Leu99 and are therefore not bound by ConA. We also investigated one other complex containing a pyranoside ring, methyl α-mannoside 10. According to our calculations there is a small increase in the difference in binding energies (ΔEb) between 10 and 9 (0.7 kcal mol−1 and 0.1 kcal mol−1 for the two computational models considered) in qualitative agreement with the experimental increase in the enthalpy of binding.
Fig. 8A shows that 10 is bound by ConA in a very similar way as 9, maintaining the same number and type of hydrogen-bonds. Both methyl “manno” β-septanoside 7 (Fig. 8B) and methyl “gluco” β-septanoside 6 (Fig. 8C) are the seven-membered ring homologs of methyl α-mannoside 10 and methyl α-glucoside 9, respectively. The seven hydrogen bonds that are critical to binding pyranosides were conserved for both of the methyl β-septanoside ligands. Inspection of the components that contribute to the binding energies of manno-configured methyl glycosides 7 and 10 showed that the energy required to deform the molecule to fit in the ConA active site was the same for both molecules (Table 2). The main difference, therefore, came from different interactions with the protein. Considering the van der Waals contribution to the protein–ligand interaction energy, which is readily available in QM/MM calculations, revealed that the complex ConA·7 is about 1.5 kcal mol−1 more stable than its six-member ring counterpart (ConA·10), despite having an overall less favorable free energy of binding. Therefore, the origin of the more favorable affinity of ConA for the pyranoside must be dominated by entropy. Indeed, due to the additional stabilization of ConA·7versus ConA·10 coming from more favorable van der Waals contacts (ΔH(ConA·7) − ΔH(ConA·10) < 0), 7 is more tightly bound than 10. Thus, it is expected that 10, in the ConA·10 complex, should have a larger conformational entropy than 7 in the ConA·7 complex (TΔS(ConA·7) − TΔS(ConA·10) < 0), which is in agreement with the ITC data. On the other hand, a similar comparison of gluco-configured methyl glycosides 6 and 9 revealed that 9 takes considerably less energy to adopt the bound conformation than does 6. Such an enthalpic difference is what finally dominates, resulting in larger binding affinities for the complex ConA·9 with respect to ConA·6, which is also in agreement with the ITC data.
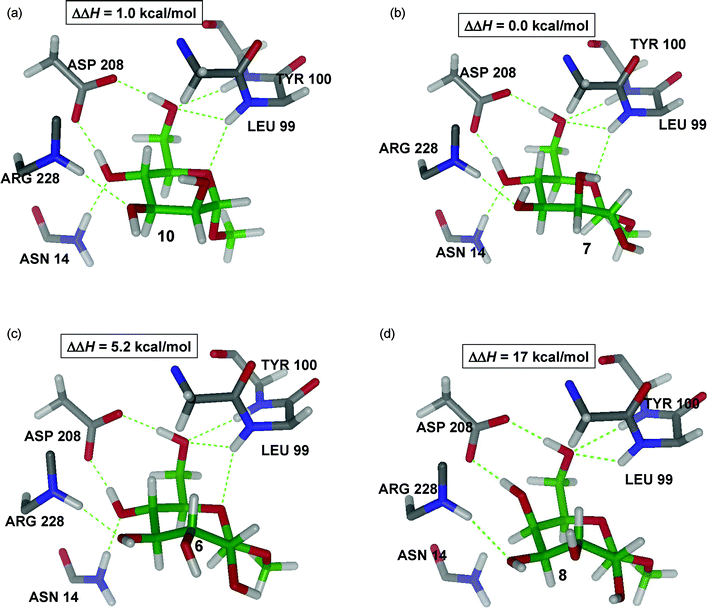 | ||
| Fig. 8 QM/MM structures of complex ConA with ligands 6, 7, 8, and 10 showing the main interactions with the protein. | ||
We next sought to determine the origin of the different affinities between the two methyl β-septanosides. The difference of ∼ 5 kcal mol−1 in the calculated binding energy between methyl “manno” β-septanoside 7 and methyl “gluco” β-septanoside 6 comes mostly from differences in the deformation energy. It takes about 4 kcal mol−1 more energy to reconfigure ligand 6 into a conformation appropriate for binding relative to 7.
As noted previously, the conformation of unbound 6 is the same as we had determined in our earlier conformational analysis.36 We argue that, based on this correspondence, the conformation of free ligand 7, following the present minimization protocols, is also accurate. Inspection of the pairs of structures (unbound and bound for both 6 and 7) shows that the main difference between them comes from the reorientation of the ring hydroxyl groups in going from the free to the bound state (Fig. 9). Reorientation of the ring hydroxyls is governed by the need to make the C5 and C7 hydroxyl groups available to donate hydrogen-bonds to Asp208. One way to describe these changes is to count the number of electrostatic OH⋯O interactions before and after binding. In methyl “manno” β-septanoside 7, this number changes from three to two (see arrows in Fig. 9), while in methyl “gluco” β-septanoside 6 it changes from five to three, accounting for most of the difference in binding energy between these two ligands. A similar analysis can be invoked for ligand 15 (figure shown in Supporting Information), which has the same stereochemistry as 6, but contains a methyl ether rather than a hydroxyl group at C2.
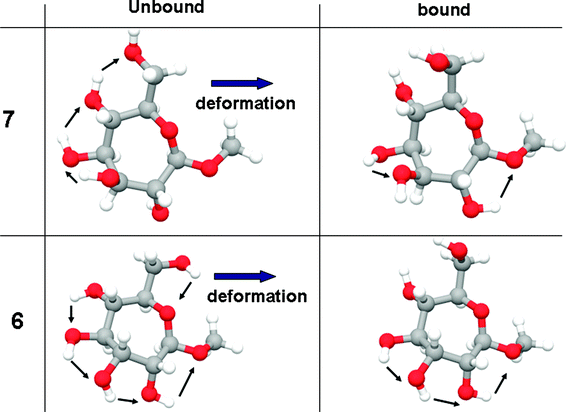 | ||
| Fig. 9 Changes in the number of internal OH–O interactions upon deformation to fit the active site. | ||
Non-binding ligands
A few of the ligands were either not bound by ConA (i.e.8, 14) or were only very weakly bound (i.e.12, 13) as measured by ITC. The low stability of the ConA·8 complex (Fig. 8D) clearly comes from the combined effect of breaking the hydrogen bonds between the ligand and Asn14 and Leu99 and the excessive energy required to induce fitting. As in the deformation of 6, two favorable electrostatic OH⋯O hydrogen-bonding interactions in 8 are lost in going from the unbound to the bound conformation. Additionally, an unfavorable dipole–dipole interaction is induced by the two hydroxyl groups at C2 and C4 (Fig. 8D) which are pointing in different directions. The inability of methyl “galacto” β-septanoside 8 to be bound by ConA parallels the behavior of methyl α-galactoside 11. This observation reinforced the importance of the stereochemistry of the ring from C3–C6 (septanoside numbering) for productive protein–septanoside interactions. A similar argument can be applied to the ConA·5 complex. The QM/MM structure of this complex reveals the seven hydrogen bonds between ligand and protein are conserved (figure and analysis shown in Supporting Information). Thus, the only contributor to destabilization comes from a large deformation energy upon binding titrations of ConA with methyl “manno” α-septanoside 14 also did not show evidence of binding. Whereas the stereochemistry at C5 in 8 prevented binding, it was the anomeric (C1) configuration of 14 that obstructed protein association. Methyl septanosides 7 and 14 share the same stereochemistry around the ring from C2–C6; the only difference between them is that 7 is a methyl β-septanoside and 14 is a methyl α-septanoside. Results obtained were consistent with our earlier observation that α-septanosides were not ligands of ConA. This reversal of selectivity (β-anomers bound in preference to α-anomers) for septanoside binding relative to pyranosides (α-anomers bound in preference to β-anomers) was attributed to a combination of septanoside conformation in the bound state and the interaction between the aglycone methyl group and the Leu99 side chain.The configuration of stereocenters in oxepanes 12 and 13 is consistent with that of glucose and mannose, respectively. Titration data for oxepanes 12 and 13 suggested that they may be ligands of ConA but, if so, their affinities were below those reported for the weakest binders (6 and 15) in Table 1. These results reinforced the fact that the anomeric center was a necessary feature for binding and that it likely aided in rigidifying the structure of the septanoside. We propose that these oxepanes are in fact bound by ConA, but with very low affinity. Glycerol, for example, makes contacts to ConA in the monosaccharide binding pocket, as evidenced in X-ray structures.49 Overall, the ligands that demonstrated low or no affinity for ConA were essential to the development of a general model of how structural features affected affinity Table 2.
Conclusions
The observation and analysis of ConA binding to monosaccharides reported here leads to three major conclusions: first, methyl β-septanosides are bound by ConA in a manner that is analogous to methyl α-pyranosides. Second, the greater binding affinity of pyranosides versus septanosides has a different origin for glucosides and mannosides; in glucosides, the lower affinity of the septanosides is dominated by an unfavorable enthalpy contribution, while in mannosides, the lower affinity of the septanoside is dominated by an unfavorable conformational entropy. Third, the hypothesis of a constant solvent reorganization enthalpy amongst the ligands considered has been confirmed.An important contributor to the differential binding enthalpy among natural and unnatural monosaccharides is the energy required to reorient the hydroxyl groups of a given ligand. This is mainly the result of the loss of two internal contacts involving the hydroxyls, which are always needed to bind Asp208. Since the stereochemistry is different among many of these ligands, this reorientation is different among them. The two non-binding events found experimentally and computationally were rationalized in terms of an unfavorable enthalpic contribution due to both the deformation energy and the loss of intermolecular hydrogen bonds with the protein when complexed.
Experimental section
Provided here are schemes and procedures for the preparation of molecules 12, 13, and 15. Characterization data used to confirm structures of intermediates and products is provided in the supporting information.Unless other conditions were specified, reactions were conducted at room temperature under nitrogen atmosphere and all the solvents and reagents were purchased commercially and used without further purification. Reactions were monitored by TLC (silica gel, 60 Å, F254, 250 nm) with visualization conducted either under UV light or by charring with 2.5% p-anisaldehyde in H2SO4, acetic acid, and ethanol solution. Flash column chromatography was conducted on silica gel (60 Å, 32–63 μm). Optical rotations were measured at 22 ± 2 °C. 1H NMR spectra were recorded at 300, 400 and 500 MHz with chemical shifts referenced to the residual signal in CDCl3 (δH 7.27 ppm) or CD3OD (δH 3.31 ppm). 13C spectra were collected at 75, 100 and 125 MHz and referenced to the residual signal in CDCl3 (δC 77.2) or CD3OD (δC 49.0 ppm).
 | ||
| Scheme 1 Preparation of oxepanes 12 and 13. | ||
1,6-Anhydro-2-deoxy-D-glycero-D-glucoseptitol (12)
20% Pd(OH)2/C (0.106 g) was added to a solution of oxepine 16 (0.112 g, 0.209 mmol) in MeOH (7 mL). The reaction vessel was purged with N2, then H2 was introduced via a balloon attachment. The mixture was allowed to stir overnight (15 h) at rt. The vessel was purged with N2 before being exposed to the atmosphere. The mixture was then filtered through a short pad of celite. The celite was washed with MeOH (4 × 5 mL) and the solvent was removed from the combined filtrates by rotary evaporation under reduced pressure to give 1,6-anhydro-2-deoxy-D-glycero-D-glucoseptitol (12) as a beige solid (0.035 g, 99%). [α]D −12.62 (c 1.96, CH3OH); 1H NMR (CD3OD) 500 MHz δ 3.77–3.71 (m, 1H), 3.69 (dd, J = 11.0, 2.8 Hz, 1H), 3.61 (ddd, J = 12.1, 5.2, 4.6 Hz, 1H), 3.56–3.51 (m, 1H), 3.41 (dd, J = 11.6, 7.0 Hz, 1H), 3.24 (ddd, J = 9.3, 6.8, 2.8 Hz, 1H), 3.18–3.10 (m, 2H), 1.82–1.78 (m, 2H); 13C NMR (CD3OD) 75 MHz δ 82.3, 79.3, 74.9, 71.2, 66.4, 64.4, 34.3; HRMS [M + Na]+m/z calcd for C7H14O5Na 201.0733, found 201.0727.1,6-Anhydro-2-deoxy-D-glycero-D-mannoseptitol (13)
20% Pd(OH)2/C (0.157 g) was added to a solution of oxepine 17 (0.100 g, 0.187 mmol) in MeOH (10 mL). The reaction vessel was purged with N2, then H2 was introduced via a balloon attachment. The mixture was allowed to stir overnight (15 h) at rt. The vessel was purged with N2 before being exposed to the atmosphere. The mixture was then filtered through a short pad of celite. The celite was washed with MeOH (4 × 5 mL) and the solvent was removed from the combined filtrates by rotary evaporation under reduced pressure to give 1,6-anhydro-2-deoxy-D-glycero-D-mannoseptitol (13) (0.028 g, 93%). [α]D −2.05 (c 1.00, CH3OH); 1H NMR (CD3OD) 300 MHz δ 4.10 (dddd, J = 9.0, 1.7, 1.6, 1.5 Hz, 1H), 4.02 (ddd, J = 12.4, 5.2, 5.0 Hz, 1H), 3.74–3.69 (m, 2H), 3.60–3.47 (m, 3H), 3.35–3.28 (m, 1H), 2.13 (dddd, J = 18.1, 9.1, 5.3, 5.3 Hz, 1H), 18.0–1.71 (m, 1H); 13C NMR (CD3OD) 100 MHz δ 86.0, 80.2, 72.2, 71.1, 68.1, 64.9, 34.2; HRMS [M + Na]+m/z calcd for C7H14O5Na 201.0733, found 201.0728.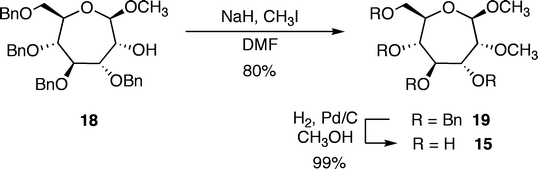 | ||
| Scheme 2 Preparation of methyl 2-O-methyl-β-septanoside 15. | ||
Methyl 3,4,6-tetra-O-benzyl-2-O-methyl-β-D-glycero-D-gulosepanoside (19)
To an ice cold solution of methyl 3,4,5,7-tetra-O-benzyl-β-D-glycero-D-guloseptanoside 18 (0.077 g 0.132 mmol) in THF (5 mL) was added sodium hydride 60% oil dispersion (0.006 g, 0.145 mmol) followed by methyl iodide (0.024 mL, 0.296 mmol) in two portions. The reaction was allowed to warm to rt and stirred for 18 h. The reaction was then quenched with the addition of 5 mL EtOAc–H2O (1:1). The mixture was concentrated and extracted with DCM (3 × 20 mL). The combined extract was dried with Na2SO4 and the solvent was removed under reduced pressure. The resulting residue was purified viacolumn chromatography using 7:3 hexanes:EtOAc as eluent to give methyl 3,4,6-tetra-O-benzyl-2-O-methyl-β-D-glycero-D-gulosepanoside (19) as a colorless oil (0.063 g, 80%). [α]D +14.79 (c 3.21, CDCl3); IR (KBr) cm−1 3087.66, 3062.98, 3030.11, 2930.80, 2862.78, 1960.11, 1872.13, 1811.78, 1722.81, 1604.74, 1496.60, 1453.47, 1387.69, 1361.08, 1229.92, 1276.26, 1206.95, 1110.10, 1074.43, 1028.00, 987.35, 910.51, 736.29, 697.40, 600.75, 484.17; 1H NMR (CDCl3) 300 MHz δ 7.40–7.20 (m, 20H), 4.72–4.54 (m, 8H), 4.40 (d, J = 11.25 Hz, 1H), 4.07 (ddd, J = 9.7, 6.4, 2.3 Hz, 1H), 4.00–3.95 (m, 2H), 3.72 (dd, J = 10.2, 2.0 Hz, 1H), 3.67–3.57 (m, 3H), 3.55 (s, 3H), 3.46 (s, 3H); 13C NMR (CDCl3) 75 MHz δ 138.7, 138.5, 138.4, 128.6, 128.5(2), 128.3, 128.1, 128.0, 127.9, 127.8, 127.6(2), 106.0, 83.0, 81.4, 79.2, 77.9, 75.8, 73.6, 73.5, 73.4, 73.2, 71.7, 59.2 56.4; FAB-MS [M + Na]+m/z calcd for C37H42O7Na+ 621.2823, found 621.2801.
Methyl 2-O-methyl-β-D-glycero-D-gulosepanoside (15)
20% Pd–OH/C (0.053 g) was added to a solution of 19 (0.062 g, 0.103 mmol) in MeOH (5 mL). The reaction vessel was purged with N2, then H2 and was kept under an H2 atmosphere via a balloon attachment. The mixture was allowed to stir 15 h at rt. The vessel was then evacuated of H2 and purged with N2 before being exposed to the atmosphere. The mixture was filtered through a short pad of celite and washed with CH3OH (4 × 5 mL). The solvent was removed from the combined filtrates by rotary evaporation under reduced pressure to give 15 as an off-white solid (0.024 g, 99%); [α]D +6.34 (c 1.30, CH3OH); 1H NMR (CD3OD) 300 MHz δ 4.48 (d, J = 3.6 Hz, 1H), 3.87 (d, J = 11.3, 2.3 Hz, 1H), 3.78–3.70 (m, 2H), 3.62–3.49 (m, 3H), 3.48 (s, 3H), 3.47 (s, 3H); 13C NMR (CD3OD) 75 MHz δ 108.0, 86.8, 79.8, 75.4, 74.8, 71.6, 64.4, 60.0, 56.6; FAB-MS [M + Na]+m/z calcd for C9H18O7Na+ 261.0945, found 261.0941.Acknowledgements
Bikash Surana (U. of Connecticut) helped with the synthesis and characterization of compound 12. Martha Morton and Srikanth Rapole (U. of Connecticut) are thanked for technical assistance with NMR and mass spectrometry, respectively. M.W.P. thanks the National Science Foundation (NSF) for a CAREER Award (CHE-0546311). J.A.G thanks financial support from the Camille and Henry Dreyfus foundation and NSF for a CAREER Award (CHE-0847340). C. V. K. and M. R. D. thank the NSF Division of Materials Research (DMR-0604815, and DMR-1005609) for partial financial support of this work.References
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- A. Imberty, H. Lortat-Jacob and S. Perez, Carbohydr. Res., 2007, 342, 430–439 CrossRef CAS.
- L. Ingrassia, I. Camby, F. Lefranc, V. Mathieu, P. Nshimyumukiza, F. Darro and R. Kiss, Curr. Med. Chem., 2006, 13, 3513–3527 CrossRef CAS.
- A. Varki, Nature, 2007, 446, 1023–1029 CrossRef CAS.
- X. L. Zhang, Curr. Med. Chem., 2006, 13, 1141–1147 CrossRef CAS.
- V. V. Glinsky and A. Raz, Carbohydr. Res., 2009, 344, 1788–1791 CrossRef CAS.
- P. Nangia-Makker, V. Hogan, Y. Honjo, S. Baccarini, L. Tait, R. Bresalier and A. Raz, J. Natl. Cancer Inst., 2002, 94, 1854–1862 CrossRef CAS.
- K. Grogan, in PharmaTimes, 2010, vol. February 3 Search PubMed.
- J. C. Wilson and M. von Itzstein, Curr. Drug Targets, 2003, 4, 389–408 Search PubMed.
- F. A. Quiocho, Annu. Rev. Biochem., 1986, 55, 287–315 CrossRef CAS.
- W. I. Weis and K. Drickamer, Annu. Rev. Biochem., 1996, 65, 441–473 CrossRef CAS.
- F. A. Quiocho, Trans. Am. Crystall. Asoc., 1989, 25, 23–35 Search PubMed.
- C. Clarke, R. J. Woods, J. Gluska, A. Cooper, M. A. Nutley and G. J. Boons, J. Am. Chem. Soc., 2001, 123, 12238–12247 CrossRef CAS.
- D. Gupta, T. K. Dam, S. Oscarson and C. F. Brewer, J. Biol. Chem., 1997, 272, 6388–6392 CrossRef CAS.
- R. Kadirvelraj, B. L. Foley, J. D. Dyekjaer and R. J. Woods, J. Am. Chem. Soc., 2008, 130, 16933–16942 CrossRef CAS.
- C. P. Swaminathan, N. Surolia and A. Surolia, J. Am. Chem. Soc., 1998, 120, 5153–5159 CrossRef CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- M. A. Boone, F. E. McDonald, J. Lichter, S. Lutz, R. Cao and K. I. Hardcastle, Org. Lett., 2009, 11, 851–854 CrossRef CAS.
- S. Castro, M. Duff, N. L. Snyder, M. Morton, C. V. Kumar and M. W. Peezuh, Org. Biomol. Chem., 2005, 3, 3869–3872 RSC.
- A. Tauss, A. J. Steiner, A. E. Stutz, C. A. Tarling, S. G. Withers and T. M. Wrodnigg, Tetrahedron: Asymmetry, 2006, 17, 234–239 CrossRef CAS.
- Z. Derewenda, J. Yariv, J. R. Helliwell, A. J. Kalb, E. J. Dodson, M. Z. Papiz, T. Wan and J. Campbell, EMBO J., 1989, 8, 2189–2193 CAS.
- S. J. Hamodrakas, P. N. Kanellopoulos, K. Pavlou and P. A. Tucker, J. Struct. Biol., 1997, 118, 23–30 CrossRef CAS.
- S. J. Harrop, J. R. Helliwell, T. C. M. Wan, A. J. Kalb, L. Tong and J. Yariv, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 143–155 CrossRef.
- P. N. Kanellopoulos, K. Pavlou, A. Perrakis, B. Agianian, C. E. Vorgias, C. Mavrommatis, M. Soufi, P. A. Tucker and S. J. Hamodrakas, J. Struct. Biol., 1996, 116, 345–355 CrossRef CAS.
- J. H. Naismith, C. Emmerich, J. Habash, S. J. Harrop, J. R. Helliwell, W. N. Hunter, J. Raftery, A. J. Kalb and J. Yariv, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 847–858 CrossRef.
- J. H. Naismith and R. A. Field, J. Biol. Chem., 1996, 271, 972–976 CrossRef CAS.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef.
- J. B. Corbell, J. J. Lundquist and E. J. Toone, Tetrahedron: Asymmetry, 2000, 11, 95–111 CrossRef CAS.
- S. M. Dimick, S. C. Powell, S. A. McMahon, D. N. Moothoo, J. H. Naismith and E. J. Toone, J. Am. Chem. Soc., 1999, 121, 10286–10296 CrossRef CAS.
- D. A. Mann, M. Kanai, D. J. Maly and L. L. Kiessling, J. Am. Chem. Soc., 1998, 120, 10575–10582 CrossRef CAS.
- E. K. Woller, E. D. Walter, J. R. Morgan, D. J. Singel and M. J. Cloninger, J. Am. Chem. Soc., 2003, 125, 8820–8826 CrossRef CAS.
- L. Bhattacharyya and C. F. Brewer, Eur. J. Biochem., 1989, 178, 721–726 CrossRef CAS.
- L. Bhattacharyya, S. H. Koenig, R. D. Brown and C. F. Brewer, J. Biol. Chem., 1991, 266, 9835–9840 CAS.
- D. K. Mandal, N. Kishore and C. F. Brewer, Biochemistry, 1994, 33, 1149–1156 CrossRef CAS.
- M. W. Peczuh and N. L. Snyder, Tetrahedron Lett., 2003, 44, 4057–4061 CrossRef CAS.
- M. P. DeMatteo, N. L. Snyder, M. Morton, D. M. Baldisseri, C. M. Hadad and M. W. Peczuh, J. Org. Chem., 2005, 70, 24–38 CrossRef CAS.
- S. D. Markad, S. J. Xia, N. L. Snyder, B. Surana, M. D. Morton, C. M. Hadad and M. W. Peczuh, J. Org. Chem., 2008, 73, 6341–6354 CrossRef CAS.
- Maestro, version 9.0, Schrodinger LLC, New York, NY Search PubMed.
- Glide, version 502076, Schrodinger LLC, New York, NY Search PubMed.
- Qsite, version 7.5, Schrodinger LLC, New York, NY Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- Jaguar, version 7.5, Schrodinger LLC, New York, NY Search PubMed.
- M. C. Chervenak and E. J. Toone, J. Am. Chem. Soc., 1994, 116, 10533–10539 CrossRef CAS.
- B. D. Isbister, P. M. St. Hilaire and E. J. Toone, J. Am. Chem. Soc., 1995, 117, 12877–12878 CrossRef CAS.
- S. Ha, J. Gao, B. Tidor, J. W. Brady and M. Karplus, J. Am. Chem. Soc., 1991, 113, 1553–1557 CrossRef CAS.
- D. F. Senear and D. C. Teller, Biochemistry, 1981, 20, 3083–3091 CrossRef CAS.
- Values of c were determined using c = Ka[ConA], with Ka values of 520 and 540 M−1 and ConA values of 0.200–0.300 mM.
- W. B. Turnbull and A. H. Daranas, J. Am. Chem. Soc., 2003, 125, 14859–14866 CrossRef CAS.
- F. J. Lopez-Jaramillo, L. A. Gonzalez-Ramirez, A. Albert, F. Santoyo-Gonzalez, A. Vargas-Berenguel and F. Otalora, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1048–1056 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all newly reported compounds. Computed QM/MM structures not shown in the main text are provided. Further details on the thermodynamic cycle used in our calculations and approximations are also provided. See DOI: 10.1039/c0ob00425a |
| This journal is © The Royal Society of Chemistry 2011 |