Dramatic increase of quench efficiency in “spacerless” dimaleimide fluorogens†
Karine
Caron
,
Virginie
Lachapelle
and
Jeffrey W.
Keillor
*
Département de chimie, Université de Montréal, CP 6128, Succursale centre-ville, Montréal, Québec H3C 3J7, Canada. E-mail: jw.keillor@umontreal.ca
First published on 10th November 2010
Abstract
In this post-genomic era, new techniques are needed to cope with the task of assigning functional roles to the huge number of identified putative gene products. We have developed a minimalist labelling strategy based on the use of synthetic fluorogenic probe reagents that fluoresce only after their reaction with a target peptide sequence. The probe reagents have fluorescent cores and bear two maleimide groups, such that their latent fluorescence is quenched by a photoinduced electron transfer (PET) to the pendant maleimide groups, until both of these groups undergo a specific thiol addition reaction. The efficiency of the fluorescence quenching is critical to the practicality of this labelling method, and has been predicted to be related to the intramolecular distance between the fluorophore and the maleimide groups. We have conducted the first direct test of this hypothesis by preparing a series of novel fluorogens that differ only by the spacer moiety separating their coumarin fluorophore and their dimaleimide fragment. A striking correlation was observed between intramolecular distance and the fluorescence enhancement (FE) observed after reaction with two equivalents of thiol. Guided by this observation, we then designed ‘spacerless’ fluorogens, of which a dansyl derivative shows an FE ratio of >300, the largest recorded for dimaleimide fluorogens. The trends observed herein provide valuable lessons for subsequent fluorogen design, and the novel fluorogens developed in the course of this study are currently being applied to protein labelling applications.
Introduction
The 20th century has witnessed the emergence of modern genomics, allowing the sequencing and study of the genomes of entire organisms, leading to the characterization of a considerable number of gene products. The fluorescent labelling of proteins has proven to be a powerful approach to accomplish this task in the sense that it allows the monitoring of the dynamic processes of their synthesis and degradation as well as their localization and interactions with different proteins.1–3In general two approaches have been taken in the fluorescent labelling of specific proteins. The first involves the genetic fusion of the protein of interest with another protein – one that is either intrinsically fluorescent, or an enzyme that can be labelled with a fluorescent irreversible inhibitor. For example, fusion with jellyfish green fluorescent protein (GFP) has seen particularly broad application,2,4 as has the fusion of enzymes such as phosphopantetheine transferase (PPTase),5,6O6-alkylguanine-DNA alkyltransferase (AGT)7–9 or mutant haloalkane dehalogenase (HALO).10 In all these cases, however, the steric bulk of the pendant protein may affect the native biology of the labelled protein of interest.
The second general approach involves the fusion of a short peptidic tag to the protein of interest that can either be modified enzymatically or react specifically with fluorescent labelling agents. Transglutaminase, 11biotin ligase,12lipoic acid ligase,13 and proteinfarnesyl transferase14 are examples of enzymes used in this way, while the FlAsH labelling agents15,16 represent an example of the small molecule-based labelling of a short peptide tag. This labelling method has seen widespread application, although some drawbacks have been noted, including the inherent toxicity of the organoarsenic compounds employed17 and persistent background staining.18
In order to address some of the disadvantages of existing methods, we have developed a complementary protein labelling technique based on the fluorogenic addition of a dimaleimide probe to a short α-helical sequence fused to the protein of interest.19,20 The latent fluorescence of the probe is restored upon reaction of both maleimide groups21 with the two cysteine residues, leading to the fluorescent labelling of the protein bearing the target peptidic sequence (Scheme 1). Thus far, this method has shown very low cytotoxicity and good selectivity, both representing important advantages over existing complementary methods.17,18
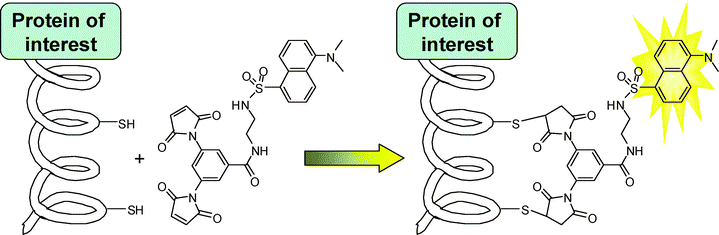 | ||
| Scheme 1 | ||
Although maleimide derivatives have been used for several years as fluorescent thiol-reactive probes,22–24 little attention has been given to their mechanism of fluorescence quenching. Our original approach to the preparation of such compounds involved the linear synthesis of a series of aromatic compounds bearing two maleimide groups attached directly to the fluorescent core.19 The quench efficiencies measured for these compounds were excellent, but their low quantum yields and difficult preparation limited their practical application. Subsequently, a second generation of dimaleimide fluorogens was prepared by a convergent synthetic strategy whereby a dimaleimide moiety is linked to a fluorophore via an intervening spacer.20,21 One of the resulting fluorogens (1, Chart 1) was used in a photophysical study that demonstrated that maleimide-mediated quench proceeds via a photoinduced electron transfer (PET) mechanism (Scheme 2).21 According to this mechanism, the intramolecular transfer of an electron from the excited state of the fluorophore to the LUMO of the maleimide group results in attenuated fluorescence due to non-radiative relaxation. However, after the maleimide group undergoes its well-known thiol addition reaction, the LUMO of the resulting succinimide group is higher in energy, and electron transfer from the fluorophore excited state is no longer thermodynamically favoured. As a result, relaxation occurs directly to the ground state with accompanying fluorescent emission.21
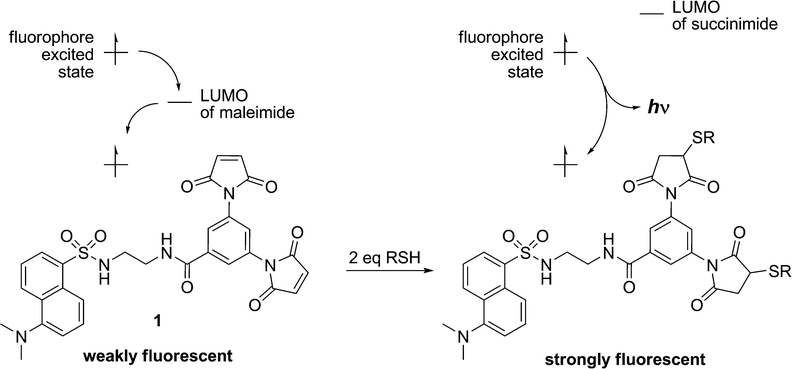 | ||
| Scheme 2 Fluorescence quenching by Photoinduced Electron Transfer (PET). | ||
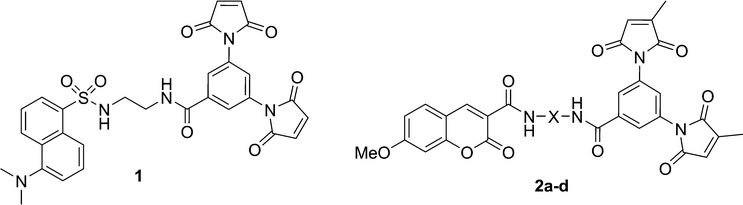 | ||
| Chart 1 | ||
The practicality of the application of this fluorogenic reaction to the fluorescent labelling of proteins is directly related to the efficiency of the fluorescence quench. Fluorogens that are efficiently quenched display lower background fluorescence, allowing one to envisage a protein labelling technique that does not require washing to remove excess labelling agent. To this end, we sought to study and ultimately improve the efficiency of the quench reaction. The electron transfer from the fluorophore to the maleimide group that is critical to the quench mechanism21 can occur through the intervening covalent bonds or through space. Previous studies have shown that within a certain distance, through-space electron transfer is more efficient and the predominant manifold.25 Therefore, the role of the spacer moiety in the tripartite design of our fluorogens becomes crucial, in conferring a conformation wherein the pendant fluorophore and maleimide groups are in close proximity to each other. Herein, we report the synthesis of a series of coumarin-based dimaleimide fluorogens (2a–d, Chart 1), where the central spacer moiety was systematically varied, allowing comparison between fluorogen conformation and contrast ratio.
Results & discussion
Design and synthesis of fluorogens
As mentioned above, a convergent synthetic strategy for the preparation of dansyl fluorogens was conceived in a previous study and shown to be an efficient way to provide access to series of fluorogens.21 In more recent work, we have shown that the 3-methylmaleimide group allows for better kinetic discrimination between a target protein and adventitious reaction with small molecule thiols such as glutathione.20 Therefore, the dimaleimide fragment used in this study was designed to bear two such 3-methylmaleimide groups. As shown in Scheme 3, dimaleimide fragment 4 was prepared from 3,5-diaminobenzoic acid and citraconic anhydride. Since our previously published procedure20 we have optimised conditions for the synthesis of this fragment. In particular, the maleimide ring closure can be effected reproducibly and with excellent yield in the presence of ZnCl2 and HMDS (Scheme 3) rather than with the commonly used acetic anhydride and sodium acetate.20,26–28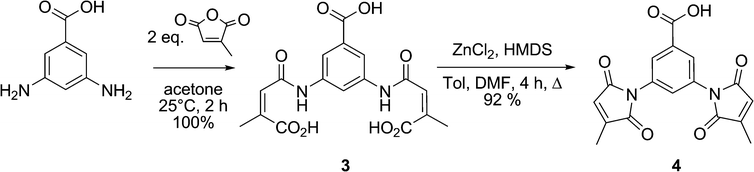 | ||
| Scheme 3 Synthesis of dimaleimide moiety 4. | ||
Coumarin was chosen as the fluorophore in the present series of fluorogens for a number of reason, both synthetic and photophysical. For this study, the blue fluorescence of coumarin was deemed to be useful, since the relatively high energy of the corresponding excited state signifies that electron transfer to the maleimide LUMO will be thermodynamically favoured, according to the Rehm–Weller equation.29 Although coumarin derivatives are well-known and have been used in fluorescent probes, their syntheses can be laborious and may suffer from low yields. Moreover, few coumarin derivatives are characterised by high quantum yields. One exception is 3-carboxy-7-methoxycoumarin (5), whose electron donating group at position 7 and electron withdrawing group at position 3 are apparently responsible for its intense fluorescence (ΦF = ∼0.8 (H2O)).30 Furthermore, efficient synthetic routes to these particular coumarin derivatives have been developed, including the method we employed, namely, the condensation of Meldrum's acid with 4-methoxysalicylaldehyde,31 shown in Scheme 4.
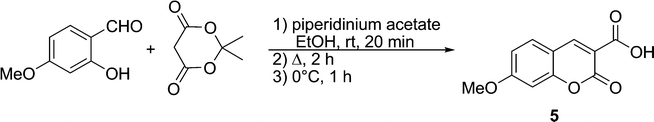 | ||
| Scheme 4 Synthesis of coumarin fluorophore 5. | ||
Dimaleimide fragment 4 was then linked to 3-carboxy-7-methoxycoumarin (5), via a series of diamine spacers. In our previous photophysical study, ethylenediamine was used as a short but conformationally flexible linker in the tripartite design. This served as a point of reference for the spacers used in the present work, whereas other diamine spacers were chosen to allow control over the distance between the fluorophore and dimaleimide moieties. The monoprotection of ethylenediamine (as 6a) can be accomplished in excellent yield using a ten-fold excess of diamine over di-tert-butyl carbonate (Boc2O). However, this approach is impractical for the other, more expensive, diamines used in this study. Therefore, the latter diamines were monoprotected by slow addition of one equivalent of di-tert-butyl carbonate to a dilute solution of diamine, giving compounds 6b–d in fair to good yields. Fluorophores 7a–d were then prepared in a convergent manner by coupling fluorophore 5 to spacers 6a–d in the presence of EDC, HOBt and NEt3, as shown in Scheme 5. After removal of the Boc group with an excess of TFA, subsequent coupling with dimaleimide moiety 4 under the same conditions gave fluorogens 2a–d.
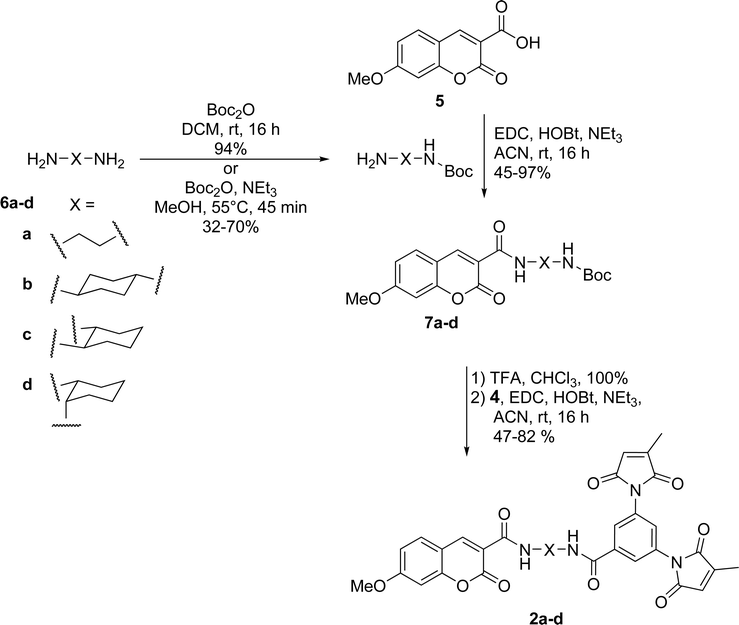 | ||
| Scheme 5 Synthesis of coumarin fluorogens 2a–d. | ||
Compounds 2a–d incorporate, as spacers, the simplest diamines that are commercially available for the practical synthesis of fluorogens according to our previously published tripartite design.21 In order to study labelling agents having even shorter fluorophore–maleimide distances, we were obliged to significantly alter our fluorogens. For example, the spacer moiety could be eliminated altogether if the dimaleimide fragment were re-designed to present an amine group, to which a fluorophore could be attached directly. Initially we envisioned designing an aniline-based dimaleimide fragment and attaching it directly to coumarin 5. However, it has been shown previously that the quantum yield of coumarin derivatives bearing a phenyl ring via a conjugated amide group at position 5 suffer from drastically reduced quantum yields.32 For this reason, a benzylamine-based dimaleimide fragment was designed instead.
As shown in Scheme 6, 3,5-dinitrobenzyl alcohol 8 was prepared by reduction of the corresponding carboxylic acid and subsequently converted into protected amine 9 by using phthalimide as a nucleophile under Mitsonobu conditions. Subsequent exchange of protecting groups gave Boc-protected amine 10. Treatment of 10 with TFA and coupling of the resulting free amine with 7-methoxycoumarin-3-carboxylic acid resulted in coumarin derivative 11, which was then submitted to catalytic hydrogenation. Diamine 12 was then allowed to react with citraconic anhydride leading to the dimaleimide fluorogenic probe 13.
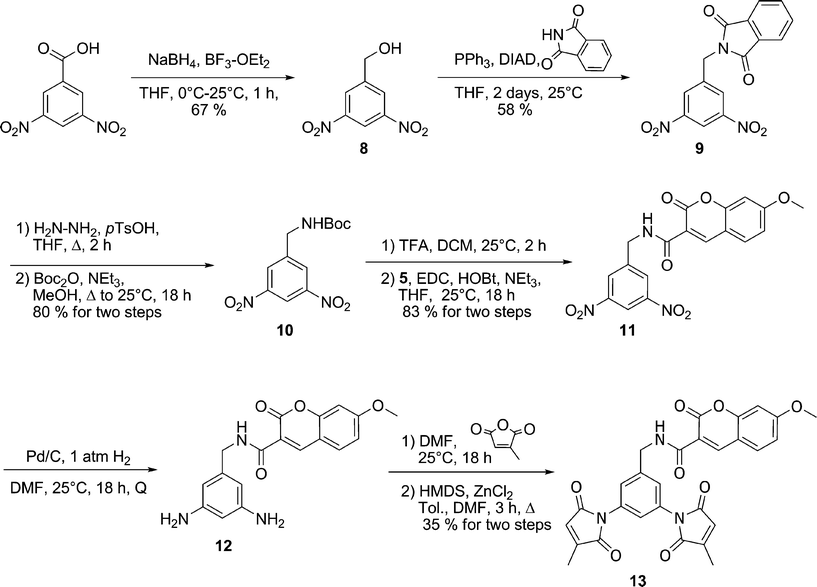 | ||
| Scheme 6 Synthesis of benzyl coumarin fluorogen 13. | ||
Quench efficiency
The effects of the spacer moieties in fluorogens 2a–d and 13 were then determined by measurement of the increase in fluorescence intensity upon reaction with two equivalents of thiol. The conditions for this fluorogenic reaction were first optimised. The hydrophilic thiol 2-mercaptopropionic acid (MPA) was chosen, owing to the practical convenience of its low volatility and aqueous solubility. Given the potential application of these fluorogens as protein labelling agents, their reaction with MPA (Scheme 7) and the accompanying fluorescence measurements was studied in a pertinent aqueous solvent mixture (5% DMSO in 50 mM HEPES buffer (pH 7.4)). The appropriate fluorogen concentration for these measurements was also determined by measuring the range over which the fluorescence was linear, namely, up to 5 μM. At this concentration it was then determined that the reaction with 50 eq of MPA at room temperature is complete after 6 h. High resolution mass spectroscopy was used to confirm that the final addition product was the expected dithiol adduct.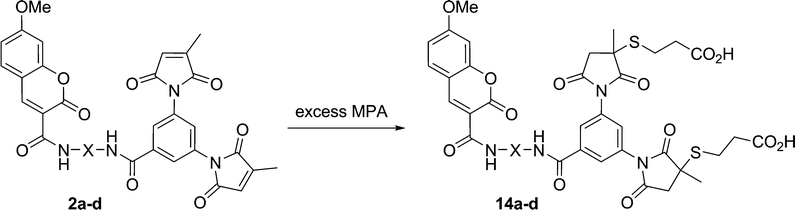 | ||
| Scheme 7 Fluorogenic thiol addition reaction between coumarin fluorogens and MPA. | ||
Measurement of fluorescence intensity before and after reaction with excess MPA provided contrast ratios, reported as the fluorescence enhancement (FE) of each fluorogen in Table 1. Since little change in excitation or emission wavelengths (λexc = 347 nm; λem = 404 nm) was observed over the course of the fluorogenic reaction, it is not surprising that little difference was observed between FE values based on either fluorescence peak height or peak area.
|
|
||||
|---|---|---|---|---|
| Fluorogen | Y | FE a | Through-space distance b/Å | Through-bond distance/Å |
| a Fluorescence enhancement, based on the ratio of fluorescence intensity (peak height) measured at λexc = 347 nm and λem = 404 nm, in 50 mM HEPES buffer (pH 7.4) containing 5% DMSO. b Mean ± standard deviation. | ||||
| 2a |
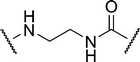
|
3.17 | 15.6 ± 0.9 | 19.9 |
| 2b |
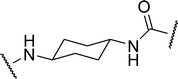
|
1.84 | 16.8 ± 1.5 | 22.9 |
| 2c |
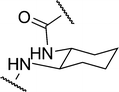
|
5.90 | 14.1 ± 2.4 | 19.9 |
| 2d |
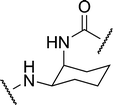
|
4.76 | 13.4 ± 1.4 | 19.9 |
| 13 |
|
6.40 | 11.8 ± 1.5 | 15.5 |
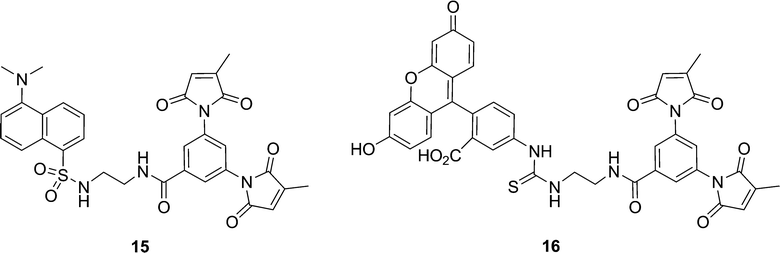
In general, the FE values determined for the series of coumarin fluorogens 2a–d are comparable with those reported previously, for similar dansyl and FITC-based fluorogens. For example, fluorogen 2a, having an ethylenediamine spacer, can be compared to recently reported dansyl fluorogen 15 and FITC fluorogen 16 (Chart 2). Fluorogen 2a displays a fluorescence enhancement (FE) value of 3.17 (Table 1) whereas dansyl fluorogen 15 has an FE of 2.0 and FITC fluorogen 16 has an FE of 5.8.20 However, while all three of these fluorogens may have the same spacer, they possess different fluorophores. This means the differences in their FE values may be as dependent on the different energies of their excited states and the thermodynamics of electron transfer to the pendant maleimide group, according to the Rehm-Weller equation,29 as they are on conformational effects25 within the fluorogens. Therefore, the series of fluorogens studied herein was prepared in order to be able to study the effect of spacer conformation alone.
 | ||
| Chart 2 | ||
A spacer that links a fluorophore and a pendant maleimide group can be thought to affect quench efficiency principally by the distance that it confers between the two moieties.21,29 While the photoinduced electron transfer (PET) responsible for fluorescence quenching can occur either through bonds or through space, the latter is considered to be the more effective manifold for small molecules such as the fluorogens studied herein.29 Molecular modelling was therefore used to predict the maleimide-to-fluorophore distance of the coumarin–dimaleimide fluorogens studied herein. In order to evaluate the effective contribution of each of the two maleimide groups to quenching as realistically as possible, the through-space distance for each maleimide group should be determined separately, within a large population of potential conformers. Therefore, molecular dynamic calculations were used to simulate how the conformation of each fluorogen varies over time. Each of the 11000 molecular dynamic snapshots obtained over the course of a 110 ps run was then minimized semi-empirically (AM1), and the distances between the coumarin fluorophore and both maleimide groups were measured. The time-averaged fluorophore–maleimide distance was then calculated for each fluorophore from the theoretical data. As shown in Table 1, these through-space distances vary significantly, by up to ∼5 Å within the series of fluorogens studied, around an average spacer length of ∼14 Å.
The effect of this variation of through-space distance on the measured FE values is equally evident. Also shown in Table 1, the measured FE values were found to vary between <2 and >6. Furthermore, these values correlate roughly linearly with the through-space distances calculated between the maleimide groups and the pendant coumarin fluorophore (Fig. 1). It is particularly instructive to consider the results obtained with benzyl derivative 13. Although it lacks the same diamine spacer structure of compounds 2a–d, it still possesses the same fluorophore and dimaleimide moieties, and its FE demonstrates the same distance dependence. This is consistent with the hypothesis that fluorophore-quencher distance is a critical parameter for quench efficiency among fluorogens comprising the same of each of these two moieties. By way of comparison, the average through-bond distance between maleimide and fluorophore was also determined, from the minimized structure of each fluorogen. Although a similar general trend may be inferred from the through-bond data also shown in Fig. 1, they are clearly not as closely correlated as the through-space distance data. This observation is consistent with the theory that through-space electron transfer is the dominant electron transfer manifold for PET quenching in such small molecule fluorogens.29
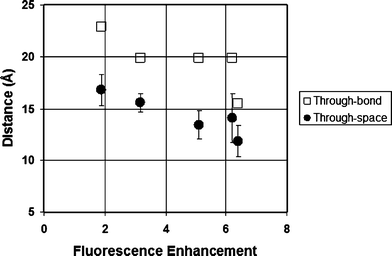 | ||
| Fig. 1 Correlation between fluorescence enhancement and maleimide–fluorophore distance (see Table 1). | ||
Spacerless dansyl fluorogen
Based on the results obtained for the series of coumarin fluorogens, we designed another ‘spacerless’ fluorogen, designed to show even greater fluorescence enhancement. First, a dimaleimide aniline moiety was designed, in order to allow its direct attachment to a fluorophore without an intermediate spacer moiety. Dansyl was chosen as an appropriate fluorophore, owing to the synthetic convenience of the sulfonamide coupling reaction and the potential to make comparisons with previously prepared dansyl fluorogens.20,21 As shown in Scheme 8, the synthesis of the dimaleimide aniline fragment was initiated with a Schmidt rearrangement, leading quantitatively to 3,5-dinitroaniline (17). This was then submitted to catalytic hydrogenation and a mono Boc protection, giving triamine 18 as a base scaffold for the new dimaleimide fragment. This mono-protected phenylenetriamine was then reacted with excess citraconic anhydride, leading to the dimaleamic acid derivative which was then cyclised to the dimaleimide core (19) upon treatment with HMDS and ZnCl2. The dansyl aniline fluorogen (21) was then obtained by deprotection of the Boc group with excess TFA and coupling of the corresponding aniline with dansyl chloride in pyridine.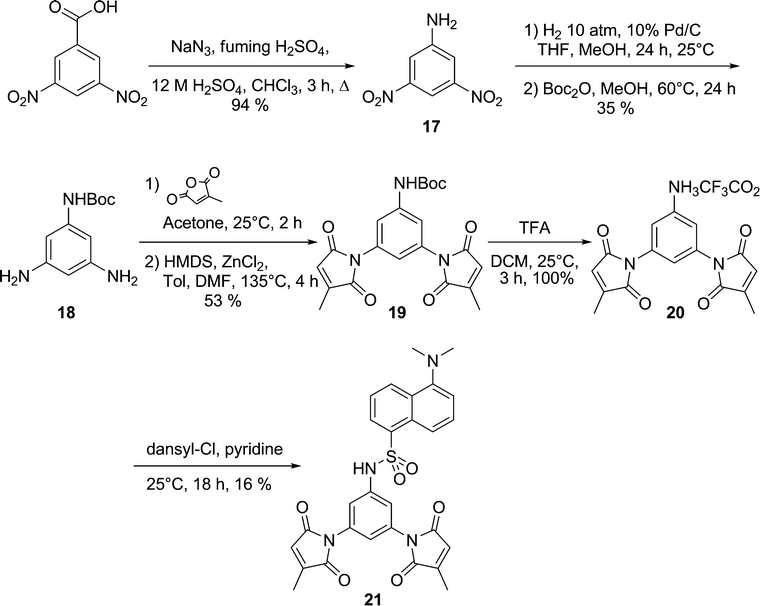 | ||
| Scheme 8 Synthesis of spacerless dansyl fluorogen 21. | ||
The FE value of fluorogen 21 was then measured as described above for the coumarin series. We were pleased to observe an increase in fluorescence intensity of at least two orders of magnitude upon thiol addition (Fig. 2). The quench efficiency in fluorogen 21 is so high, that on a practical level, it is difficult to distinguish its low fluorescence from background noise, leading to significant error in the calculation of its FE. However, by a conservative estimate, its fluorescence intensity increases by a factor of more than 300-fold upon reaction with MPA.
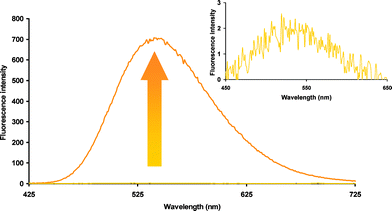 | ||
| Fig. 2 Fluorescence increase upon reaction of fluorogen 21 with two equivalents of MPA. | ||
Presumably, the extraordinary quench efficiency inherent in fluorogen 21 is due to its unique structure. Of course, its ‘spacerless’ design brings the dimaleimide and dansyl moieties into close proximity, but the conformation of the sulfonamide linkage probably plays an important role, as well. When molecular modelling was performed on 21 as for the other fluorogens presented above, the minimised structure, displayed in Fig. 3, suggests that the sulfonamide group confers a sharp turn, characterised by an acute C–S–N–C torsion angle of ∼75°. Furthermore, conjugation of the aromatic NH group presumably limits rotation around the C–N bond, resulting in the placement of a maleimide group over the dansyl fluorophore, within <7 Å of its core. Although comparison of this distance–FE correlation with those shown in Fig. 1 is not justified, since the fluorophores are different, it is obvious that the FE ratio of 21 is at least an order of magnitude greater than what we would have roughly predicted from Fig. 1. Specific orbital interactions that facilitate fluorescence quenching may be favoured in the most highly populated conformation of 21. The discovery of such a highly efficient fluorogen has opened the door to a variety of protein labelling applications as well as the design of future generations of fluorogens.
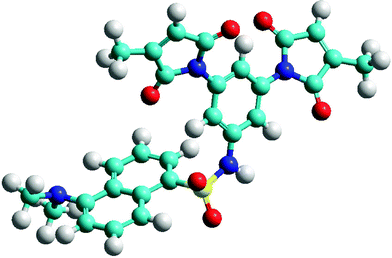 | ||
| Fig. 3 Minimised structure of fluorogen 21, showing proximity of dansyl fluorophore to maleimide group. | ||
Conclusions
The effect of spacer length and conformation on fluorescence quenching, studied in the series of coumarin maleimides presented above, confirmed the importance of through-space distance in the design of efficient fluorogens. This led to the design of dimaleimide benzyl amine and aniline derivatives that were coupled directly to coumarin and dansyl fluorophores, respectively. The resulting dansyl fluorogen 21 is the most efficiently quenched dimaleimide fluorogen known and the utility of the dimaleimide aniline moiety is currently being explored, in conjunction with other fluorophores, for sensitive protein labelling applications. Furthermore, these fluorogens offer another aspect for potential exploitation. Considering the two-point attachment to a target protein conferred by the dimaleimide moiety, a conformationally constrained fluorogen would allow for the covalent labelling of a protein with a fluorophore whose orientation is comparatively fixed, relative to the protein core. The potential application of such a labelling technique to the investigation of protein orientation and dynamics, through polarization techniques,33 is currently being explored.Experimental section
Generalities
All synthetic starting materials were purchased from Sigma Aldrich and used without further purification. Solvents were dried using GlassContour System (Irvine, CA) columns. Reactions requiring anhydrous conditions were carried out under dry nitrogen atmosphere employing conventional bench-top techniques. 13C-NMR and 1H-NMR spectra were recorded on AMXR400 or AMX300 spectrometers and were referenced to the residual proton or 13C signal of the solvent. Mass spectra were determined by FAB+ ionization on an AutoSpec Q spectrometer at the Regional Mass Spectrometry Centre at the Université de Montréal. Melting points (uncorrected) were determined on an EZ-Melt (SRS) melting point apparatus.Synthesis
1 H-NMR (300 MHz, DMSO- d 6 ) δ 7.94 (d, J = 1.8 Hz, 2H), 7.62 (t, J = 1.8 Hz, 1H), 6.83 (q, J = 1.8 Hz, 2H), 2.08 (d, J = 1.8 Hz, 6H); 13C-NMR (75 MHz, DMSO-d6)δ 170.4, 169.5, 166.2, 146.2, 132.8, 132.2, 128.2, 127.8, 126.0, 11.0; HRMS (ESI) Calculated for C17H13N2O6 [M + H]+: 341.0768. Found: 341.0770. mp 254 °C (dec)
1 H-NMR (400 MHz, DMSO- d 6 ) δ 8.72 (s, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.04 (d, J = 2.1 Hz, 1H), 7.00 (dd, J = 2.4 and 8.4 Hz, 1H), 3.89 (s, 3H); 13C-NMR (75 MHz, DMSO-d6)δ 165.9, 165.5, 158.5, 158.2, 150.4, 115.1, 114.6, 112.9, 101.5, 57.5; HRMS (ESI) Calculated for C11H8O5Na [M + Na]+: 243.0266. Found: 243.0264. mp 169 °C
1 H-NMR (300 MHz, CDCl 3 ) δ 5.09 (bs, 1H), 3.12 (dd, J = 5.8 Hz, 2H), 2.74 (t, J = 5.8 Hz, 2H), 1.61 (s, 2H), 1.38 (s, 9H); 13C-NMR (75 MHz, CDCl3)δ 156.3, 79.2, 43.3, 41.8, 28.4; MS (ESI) Calculated for C11H22N2O2 [M + H]+: 160.12. Found: 161.13.
1 H-NMR (400 MHz, CDCl 3 ) δ 4.37 (bs, 1H), 3.39 (bs, 1H), 2.21–2.28 (m, 1H), 1.99–1.89 (m, 2H), 1.69–1.66 (m, 2H), 1.41 (bs, 9H), 1.38–1.33 (m, 5H); 13C-NMR (75 MHz, CDCl3)δ 156.1, 80.0, 50.8, 49.8, 36.2, 33.0, 32.9, 29.2; HRMS (ESI) Calculated for C11H23N2O2 [M + H]+: 215.1754. Found: 215.1754.
1 H-NMR (300 MHz, CDCl 3 ) δ 4.49 (s, 1H), 3.12 (m, 1H), 1.70 (m, 2H), 1.44 (s, 9 H), 1.22 (m, 6H); 13C-NMR (75 MHz, CDCl3)δ 157.0, 80.1, 58.6, 56.4, 36.0, 33.7, 29.2, 26.0, 25.9; HRMS (ESI) Calculated for C11H22N2O2 [M + H]+: 215.1754. Found: 215.1762.
1 H-NMR (300 MHz, CDCl 3 ) δ 4.98 (s, 1H), 3.60 (s, 2H), 3.00 (m, 2H), 1.58 (m, 5H), 1.43 (s, 9H); 13C-NMR (75 MHz, CDCl3)δ 156.5, 79.8, 52.5, 50.7, 32.9, 29.2, 28.8, 23.8, 21.6; HRMS (ESI) Calculated for C11H23N2O2: 215.1754. Found: 215.1756.
1 H-NMR (400 MHz, CDCl 3 ) δ 8.94 (bs, 1H), 8.84 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 6.95 (dd, J = 2.4 and 8.8 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H), 4.98, (bs, 1H), 3.92 (s, 3H), 3.58 (q, J = 5.6 Hz, 2H), 3.38 (q, J = 6.6 Hz, 2H), 1.44 (bs, 9H); 13C-NMR (100 MHz, CDCl3)δ 164.7, 162.6, 161.5, 156.5, 155.9, 148.2, 130.8, 114.4, 113.9, 112.1, 100.1, 79.2, 55.9, 40.5, 39.7, 28.2; HRMS (ESI) Calculated for C18H23N2O6 [M + H]+: 363.1551. Found: 363.1548.
1 H-NMR (400 MHz, CDCl 3 ) δ 8.82 (s, 1H), 7.58 (d, J = 8.8 Hz, 1H), 6.93 (dd, J = 2.4 and 8.8 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H), 4.43 (bs, 1H), 3.91 (bs, 3H), 3.47 (bs, 1H), 2.09–2.07 (m, 4H), 1.62 (s, 1H), 1.45 (bs, 9H), 1.39–1.26 (m, 5H); 13C-NMR (100 MHz, CDCl3)δ 165.6, 162.6, 162.1, 157.4, 156.1, 149.0, 131.7, 115.6, 114.9, 113.2, 101.1, 80.0, 56.8, 49.7, 48.9, 32.9, 32.8, 32.3, 29.2; HRMS (ESI): Calculated for C22H28N2O6Na [M + Na]+: 439.1840. Found: 439.1841.
1 H-NMR (300 MHz, CDCl 3 ) δ 8.82 (s, 1H), 7.58 (d, J = 8.7 Hz, 1H), 6.94 (dd, J = 2.4, 8.7 Hz, 1H), 6.87 (d, J = 2.4 Hz, 1H), 5.01 (d, J = 8.1 Hz, 1H), 3.92 (s, 3H), 3.47 (m, 1H), 2.13 (m, 2H), 1.75 (m, 2H), 1.41 (m, 4H), 1.33 (s, 9H); 13C-NMR (75 MHz, CDCl3)δ 165.6, 163.3, 162.2, 157.5, 156.6, 149.0, 131.7, 115.5, 114.8, 113.2, 101.2, 79.8, 56.8, 56.1, 54.0, 33.7, 33.2, 29.1, 25.7, 25.5; HRMS (ESI) Calculated for C22H29N2O6 [M + H]+: 417.2020. Found: 417.2032. mp 141 °C.
1 H-NMR (300 MHz, CDCl 3 ) δ 9.06 (d, J = 7.6 Hz, 1H), 8.85 (s, 1H); 7.59 (d, J = 8.8 Hz, 1H), 6.94 (dd, J = 2.4 and 8.4 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 5.07 (d, J = 7.6 Hz, 1H), 4.39 (m, 1H), 3.92 (s, 3H), 1.86 (m, 1H), 1.71 (m, 2H), 1.56 (m, 5H), 1.43 (s, 9 H); 13C-NMR (75 MHz, CDCl3)δ 165.66, 162.83, 162.77, 157.47, 156.30, 149.20, 131.76, 115.64, 114.89, 113.24, 101.09, 80.16, 77.51, 77.45, 56.85, 50.02, 31.77, 30.17, 29.72, 29.22; HRMS (ESI) Calculated for C22H29N2O6 [M + H]+: 417.2020. Found: 417.2020. mp 213 °C
1 H-NMR (400 MHz, CDCl 3 ) δ 9.19 (t, J = 5.6 Hz, 1H), 8.98 (s, 1H), 7.90 (d, J = 1.6 Hz, 1H), 7.73 (bs, 1H), 7.62–7.60 (m, 2H), 6.94 (dd, J = 2.8 and 8.8 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H), 6.51 (d, J = 2.0 Hz, 2H), 3.92 (s, 3H), 3.77–3.74 (m, 2H), 3.70–3.67 (m, 2H), 2,19 (s, 6H); 13C-NMR (75 MHz, DMSO-d6) δ 171.5, 170.6, 166.1, 165.7, 163.2, 161.9, 157.5, 149.2, 147.2, 137.0, 133.9, 133,6, 132.8, 128.9, 125.8, 116.1, 114.9, 113.4, 101.5; HRMS (ESI) Calculated for C30H25N4O9 [M + H]+: 585.1616. Found: 585.1610. mp 125 °C
1 H-NMR (400 MHz, CDCl 3 ) δ 8.82 (s, 1H), 7.79 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 2.0 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H), 6.94 (dd, J = 2.4 and 8.4 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 6.51 (d, J = 1.6 Hz, 2H), 6.06 (bs, 1H), 4.02–3.99 (m, 2H), 3.92 (s, 3H), 2.19–2.17 (m, 8H), 1.48–1.41 (m, 6H); 13C-NMR (75 MHz, CDCl3)δ 170.8, 169.7, 165.9, 165.6, 162.6, 162.1, 157.4, 149.0, 146.9, 137.5, 133.4, 131.8, 128.6, 125.8, 125.6, 115.5, 114.9, 113.2, 101.0, 68.8, 56.9, 49.2, 49.0, 32.3, 26.4, 12.1, 12.0; HRMS (ESI) Calculated for C34H31N4O9 [M + H]+: 639.2086. Found: 639.2079. mp 194 °C
1 H-NMR (300 MHz, CDCl 3 ) δ 8.96 (s, 1H), 8.88 (d, J = 7.80 Hz, 1H), 7.70 (d, J = 6.9 Hz, 2H), 7.58 (d, J = 8.7 Hz, 1H), 7.45 (m, 1H), 6.85 (dd, J = 2.4 and 8.7 Hz, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.45 (m, J = 1.8 Hz 2H), 3.98 (m, 1H), 3.83 (s, 3H), 2.31 (m, 1H), 2.31 (m, 1H), 2.13 (s, 6H), 2.03 (m, 2H), 1.45 (m, 6H); 13C-NMR (75 MHz, CDCl3)δ 170.8, 169.7, 165.7, 165.4, 164.9, 162.5, 157.6, 150.5, 146.8, 137.2, 133.3, 133.2, 132.0, 128.4, 126.2, 124.4, 114.9, 113.4, 100.9, 58.2, 56.8, 53.0, 33.0, 32.7, 25.8, 25.1, 12.1; HRMS (ESI) Calculated for C34H31N4O9 [M + H]+: 639.2086. Found: 639.2087. mp 212 °C
1 H-NMR (300 MHz, CDCl 3 ) δ 9.35 (d, 1H, J = 7.8 Hz), 9.04 (s, 1H), 8.80 (s, 1H), 7.98 (m, 1H), 7.90 (d, J = 1.8 Hz, 1H), 7.63–7.60 (m, 3H), 6.99 (dd, J = 1.8 and 8.7 Hz, 1H), 6.90 (d, J = 2.1 Hz, 1H), 6.47 (bs, 2H), 4.53 (bs, 1H), 3.92 (bs, 3H), 3.11 (bs, 1H), 2.19 (bs, 6H), 2.06–1.75 (m, 8H); 13C-NMR (75 MHz, CDCl3)δ 170.8, 169.7, 166.1, 165.9, 164.5, 162.9, 157.6, 150.2, 146.8, 137.5, 133.2, 131.8, 128.4, 125.6, 123.9, 115.1, 115.1, 113.3, 101.1, 56.9, 54.7, 50.1, 30.7, 27.8, 24.8, 21.5, 12.0; HRMS (ESI) Calculated for C34H31N4O9 [M + H]+: 639.2086. Found: 639.2084. mp 165 °C
1 H-NMR (400 MHz, ((CD 3 ) 2 CO) δ 8.91 (t, J = 0.4 Hz, 1H), 8.57 (dd, J = 1.2 and 2.0 Hz, 2H), 4.93 (d, J = 5.2 Hz, 2H), 3.57 (bs, 1H); 13C-NMR (75 MHz, (CD3)2CO)δ 149.4, 148.4 (2C), 127.0 (2C), 117.6, 62.8; HRMS (ESI) Calculated for C7H5N2O5 [M − H]−: 197.0204. Found: 197.0200. mp 89 °C.
1 H-NMR (400 MHz, CDCl 3 ) δ 8.98 (t, J = 2.0 Hz, 1H), 8.61 (d, J = 2.0 Hz, 2H), 7.93–7.89 (m, 2H), 7.81–7.77 (m, 2H), 5.05 (s, 2H); 13C-NMR (75 MHz, CDCl3)δ 168.5, 168.4, 149.6, 141.3, 135.5, 132.5, 129.6, 124.7, 119.3, 41.2; HRMS (ESI) Calculated for C15H10N3O6 [M + H]+: 328.0564. Found: 328.0568. mp 171 °C
1 H-NMR (400 MHz, CDCl 3 ) δ 8.93 (bs, 1H), 8.49 (d, J = 2.4 Hz, 2H), 5.28 (bs, 1NH), 4.52 (d, J = 6.0 Hz, 2H), 1.47 (bs, 9H); 13C-NMR (75 MHz, CDCl3)δ 155.8, 148.4, 144.2, 127.1, 117.5, 80.6, 43.4, 28.1; HRMS (ESI) Calculated for C12H15N3O6 [M]−: 297.0963. Found: 297.0966. mp 94 °C.
1 H-NMR (400 MHz, DMSO- d 6 ) δ 9.41 (t, J = 5.6 Hz, 1H), 8.45 (s, 1H), 8.72 (t, J = 2.0 Hz, 1H), 8.65 (d, J = 2.0 Hz, 2H) 7.90 (d, J = 8.8 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.05 (dd, J1 = 2.4, J2 = 8.8 Hz, 1H), 4.75 (d, J = 6.0 Hz, 1H), 3.92 (s, 3H); 13C-NMR (75 MHz, DMSO-d6) δ 165.0, 162.7, 161.1, 156.8, 148.8, 148.4, 144.6, 132.1, 128.7, 117.7, 115.0, 114.1, 112.6, 100.8, 56.8, 31.2; HRMS (ESI) Calculated for C18H13N3O8Na [M + Na]+: 422.0589. Found: 422.0595. mp 233 °C.
1 H-NMR (400 MHz, DMSO- d 6 ) δ 9.20 (bs, 1NH), 8.86 (s, 1H), 7.95–7.91 (m, 2H), 7.35 (s, 1H), 7.22 (s, 1H), 7.13 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.81 (s, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.91 (s, 3H), 2.07 (s, 6H); 13C-NMR (75 MHz, DMSO-d6)δ 171.6, 170.7, 165.8, 163.0, 162.0, 157.5, 149.4, 147.1, 142.3, 133.6, 132.9, 128.8, 126.0, 124.6, 1156.0, 115.0, 113.4, 101.6, 57.5, 43.5, 12.1; HRMS (ESI) Calculated for C28H22N3O8 [M + H]+: 528.1421. Found: 528.1401.
1 H NMR (300 MHz, CDCl 3 ) δ 8.14 (t, J = 1.8 Hz, 1H), 7.64 (d, J = 1.8 Hz, 2H), 4.39 (s (br), 2H); 13C NMR (75 MHz, CDCl3)δ 151.9, 150.1, 113.1, 105.2; HRMS (ESI) Calculated for C6H4N3O4 [M − H]−: 182.0195. Found: 182.0207. mp: 113.1 °C.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to DCM–MeOH (49:1)) to give 18 (958 mg, 4.29 mmol, 35%) as an off-white solid.
:2) to DCM–MeOH (49:1)) to give 18 (958 mg, 4.29 mmol, 35%) as an off-white solid.
1 H NMR (400 MHz, CDCl 3 ) δ 6.18 (d, J = 2 Hz, 2H), 5.95 (bs, 1H), 5.73 (t, J = 2.0 Hz, 1H), 3.57 (bs, 4H), 1.50 (bs, 9H); 13C NMR (100 MHz, CDCl3)δ 152.5, 148.0, 140.1, 96.8, 96.0, 80.1, 28.2; HRMS (ESI) Calculated for C11H17N3O2Na [M + Na]+: 246.1218. Found: 246.1214.
:5 mL) before a dilute solution of HMDS (0.84 mL, 4.02 mmol) in toluene (5 mL) was added over 20 min. The resulting mixture was then heated to reflux for 4 h after which the volatiles were evaporated under reduced pressure. The resulting residue was dissolved in EtOAc and washed successively with 0.1 M HCl and saturated Na2CO3. The crude product was then purified by flash chromatography on silica gel (Tol/EtOAc 5%) giving 19 as an off-white solid (780 mg, 1.90 mmol, 53%).
1 H NMR (400 MHz, CDCl 3 ) δ 7.42 (d, J = 2.4 Hz, 2H), 7.12 (t, J = 2.4 Hz), 6.59 (bs, 1H), 6.43 (q, J = 2.4 Hz, 2H), 2.12 (q, J = 2.4 Hz, 6H), 1.47 (bs, 9H); 13C NMR (75 MHz, CDCl3)δ 170.0, 168.9, 152.2, 145.7, 139.5, 132.5, 127.4, 116.5, 114.1, 80.9, 28.1, 11.0; HRMS (ESI) Calculated for C21H21N3O6Na [M + Na]+: 434.1331. Found: 434.1323. mp 179 °C
1 H NMR (400 MHz, (CD 3 ) 2 CO) δ 9.71 (s, 1H), 8.56 (d, J = 8.4 Hz, 1H), 8.51 (d, J = 8.8 Hz, 1H), 8.39 (dd, J1 = 1.2, 7.2 Hz, 1H), 7.62–7.58 (td, J = 1.2, 7.2 Hz, 2H), 7.28–7.27 (m, 3H), 7.11 (t, J = 2.0 Hz, 1H), 6.63–6.61 (m, 2H); 13C NMR (75 MHz, (CD3)2CO)δ 171.3, 170.4, 152.8, 135.4, 134.0, 131.7, 131.5, 130.3, 130.2, 129.6, 128.7, 124.9, 119.8, 119.6, 116.6, 115.9, 46.3, 12.1; HRMS (ESI) Calculated for C28H25N4O6S [M + H]+ 545.1516. Found: 545.1489. mp 230 °C
Determination of fluorescence enhancement ratios
Absorbance spectra were recorded at 25 °C, with a Cary-100 spectrometer. Emission spectra and fluorescence intensity measurements were recorded at 25 °C with a Cary Eclipse fluorometer. Excitation and emission slits were fixed at 5 nm.To a 1 to 4 mM solution of fluorogen in DMSO was added 3-mercaptopropionic acid (50 eq). The resulting mixture was stirred at 25 °C, in the dark for 18 h after which the fluorescence intensities of a dilution (∼4 μM final) in HEPES buffer (pH 7.4) were recorded. The final fluorescence intensity was then divided by the initial florescence intensity at the same fluorogen concentration to give the fluorescence enhancement ratio.
Molecular modelling
The geometry of each fluorogen was initially optimized in vacuo, using the AM1 semi-empirical method and the Polak–Ribiere conjugate gradient algorithm, with convergence being determined by an RMS gradient of <0.1 kcal Å−1 mol−1, according to the standard parameters of the software HyperChem Professional 7.5. Molecular dynamic simulations were then performed in vacuo by heating the system from 300 K to 1000 K over a 10 ps period, followed by a 100 ps constant temperature simulation. These conditions allowed the system to explore such conformational equilibria as cis/transamide bonds and boat/chair cyclohexane rings. At each 10 fs interval, the distance was recorded between the central carbon atom C9 of the coumarin ring and the tertiary carbon atom C3 of each of the two maleimide groups. According to the orbital density calculated by the AM1 semi-empirical method, these atoms feature prominently in the fluorophore excited state and the maleimide LUMO, thereby providing a reasonable estimate of the distance of electron transfer according to the PET quench mechanism. Final conformations were optimized by subsequent minimization according to the method described above. For the calculation of the through-bond distance of each fluorogen, the lengths of the bonds between all atoms from coumarin C9 to maleimide C3 were added.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR). KC acknowledges a postgraduate bursary from the Fonds Québecois de Recherche sur la Nature et les Technologies (FQRNT) and VL acknowledges NSERC for a summer internship bursary.References
- For a recent review, see: N. Soh, Sensors, 2008, 8, 1004 Search PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509 CrossRef CAS.
- W.-K. Huh, J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman and E. K. O'Shea, Nature, 2003, 425, 686 CrossRef CAS.
- L. Vivero-Pol, N. George, H. Krumm, K. Johnsson and N. Johnsson, J. Am. Chem. Soc., 2005, 127, 12770 CrossRef CAS.
- N. Johnsson, N. George and K. Johnsson, ChemBioChem, 2005, 6, 47 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86 CrossRef CAS.
- A. Keppler, M. Kindermann, S. Gendreizig, H. Pick, H. Vogel and K. Johnsson, Methods, 2004, 32, 437 CrossRef CAS.
- A. Keppler, H. Pick, C. Arrivoli, H. Vogel and K. Johnsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9955 CrossRef CAS.
- G. V. Los and K. Wood, Methods Mol. Biol., 2007, 356, 195–208 CAS.
- C. W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542 CrossRef CAS.
- I. Chen, M. Howarth, W. Y. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99 CrossRef CAS.
- C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernández-Suárez, S. Puthenweetil and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10914 CrossRef CAS.
- B. P. Duckworth, Z. Zhang, A. Hosokawa and M. D. Distefano, ChemBioChem, 2007, 8, 98 CrossRef CAS.
- B. A. Griffin, R. S. Adams and R. Y. Tsien, Science, 2000, 281, 269.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063 CrossRef CAS.
- M. F. Langhorst, S. Genisyuerek and C. A. O. Stuermer, Histochem. Cell Biol., 2006, 125, 743 CrossRef CAS.
- K. Stroffekava, C. Proenza and K. G. Beam, Pfluegers Arch., 2001, 442, 859 CrossRef CAS.
- S. Girouard, M.-H. Houle, A. Grandbois, J. W. Keillor and S. W. Michnick, J. Am. Chem. Soc., 2005, 127, 559 CrossRef CAS.
- J. Guy, R. Castonguay, N. B. Campos-Reales Piñeda, V. Jacquier, K. Caron, S. W. Michnick and J. W. Keillor, Mol. BioSyst., 2010, 6, 976 RSC.
- J. Guy, K. Caron, S. Dufresne, S. W. Michnick, W. G. Skene and J. W. Keillor, J. Am. Chem. Soc., 2007, 129, 11969 CrossRef CAS.
- J. E. T. Corrie, J. Chem. Soc., Perkin Trans. 1, 1994, 2975 RSC.
- M. E. Langmuir, J. R. Yang, A. M. Moussa, R. Laura and K. A. Lecompte, Tetrahedron Lett., 1995, 36, 3989 CrossRef CAS.
- J. R. Yang and M. E. Langmuir, J. Heterocycl. Chem., 1991, 28, 1177 CAS.
- T. P. Le, J. E. Rogers and L. A. Kelly, J. Phys. Chem. A, 2000, 104, 6778 CrossRef CAS.
- G. Kokotos and C. Tzougrakic, J. Heterocycl. Chem., 1986, 23, 87 CAS.
- P. Y. Reddy, S. Kondo, S. Fujita and T. Toru, Synthesis, 1998, 999 CrossRef CAS.
- M. Smet, D. Corens, L. Van Meervelt and W. Dehaen, Molecules, 2000, 5, 179 Search PubMed.
- A. Gilbert and J. Baggott, Essentials of Molecular Photochemistry, CRC Press, Boca Raton, 1991 Search PubMed.
- K. Azuma, S. Suzuki, S. Uchiyama, T. Kajiro, T. Santa and K. Imai, Photochem. Photobiol. Sci., 2003, 2, 443 RSC.
- A. Song, X. Wang and K. S. Lam, Tetrahedron Lett., 2003, 44, 1755 CrossRef CAS.
- N. Gagey, M. Emond, P. Neveu, C. Benbrahim, B. Goetz, I. Aujard, J. B. Baudin and L. Jullien, Org. Lett., 2008, 10, 2341 CrossRef CAS.
- D. Zenisek, J. Steyer, M. Feldman and W. Almers, Neuron, 2002, 35, 1085 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and fluorescence spectra of synthetic intermediates. See DOI: 10.1039/c0ob00455c |
| This journal is © The Royal Society of Chemistry 2011 |