A ferrocene functionalized rotaxane host system capable of the electrochemical recognition of chloride†
Nicholas H.
Evans
and
Paul D.
Beer
*
Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44 1865 272960; Tel: +44 1865 285182
First published on 9th November 2010
Abstract
A ferrocene appended rotaxane is prepared by chloride anion templation and ring closing metathesis. Upon removal of the chloride template, the rotaxane is demonstrated to be selective for chloride over more basic oxoanions by 1H NMR spectroscopy and electrochemistry, in marked contrast to an acyclic analogue - the first example of a solution based redox-active interlocked host system capable of the electrochemical recognition of anions.
Introduction
Rotaxanes, interlocked molecules comprising of a macrocyclic component locked onto an axle by bulky stopper groups, have been at the forefront of research into molecular machine-like nanotechnological applications due to the possibilities of shuttling and/or pirouetting of their components.1 However, these molecules also possess unique topologically constrained cavities which may be exploited to selectively bind guest species for future chemical sensory purposes.2 Despite this intriguing possibility, only a few examples of designed rotaxane host systems that bind charged species in their cavities have been reported to date.3,4 Due to the challenges of recognising anions (such as their low charge densities, various geometries, high hydration energies and pH dependence),5 rotaxane cavities are promising candidates for anion binding. Our group has previously synthesised a range of solution and surface confined rotaxanes by rational use of anion templation, and have demonstrated such interlocked host systems to selectively bind anions in highly competitive solvent mixtures.4a–iTo achieve selective sensing of guests in such rotaxane hosts, appropriate reporter group functionality needs to be incorporated into the interlocked molecular framework to provide a response (either optical or electrochemical). Relatively few examples have been designed and constructed to exhibit such sensory behaviour3a,c–d,4b,d,j and only one electrochemically senses anions, when confined to a surface.4d With the aim to selectively sensing chloride electrochemically in solution, our attention has turned towards attaching the redox-active ferrocene group to a rotaxane (see Fig. 1). Ferrocene has been demonstrated to be suitable for the detection of anions in a wide range of acyclic and macrocyclic receptors.6 However, its appearances in rotaxanes as a stopper7 or as part of the macrocyclic component4d,8 are rare compared to the numerous examples of non-interlocked ferrocene anion receptors.
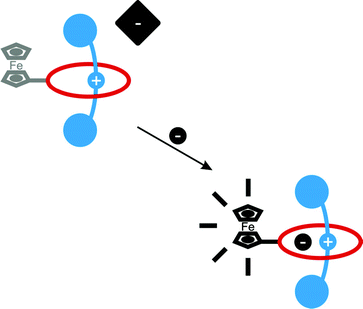 | ||
| Fig. 1 Schematic representation of the use of a ferrocene-appended rotaxane to electrochemically sense anions. | ||
In this paper we report the synthesis of the first redox-active rotaxane capable of electrochemically recognising anionic guest species, with a notably different electrochemical response to chloride compared to oxoanions being observed.
Results and discussion
Design, synthesis and characterization
To achieve the electrochemical recognition of an anion by a rotaxane, installation of an appropriate reporter group to a rotaxane capable of binding anions is required. Integration of a redox-active ferrocene moiety into such a rotaxane anion host system could be accomplished via a direct C–C bond to the isophthalamide group of an anion binding rotaxane's macrocyclic component as illustrated in Fig. 2. The chloride salt of rotaxane 1+PF6− could be prepared by a ring closing metathesis (RCM) reaction of an appropriately functionalized bis-vinyl appended macrocyclic precursor and methyl pyridinium chloride axle, the two rotaxane precursor components self-assembling in solution by a combination of anion templation, π–π stacking and hydrogen bonding.4a,c Following anion exchange rotaxane 1+PF6− contains a cavity that is anticipated to be selective for chloride,4a,c,e,i while the presence of a proximal ferrocene would enable reporting of an anion binding event by the rotaxane via cathodic shifts in the Fc/Fc+ redox couple.9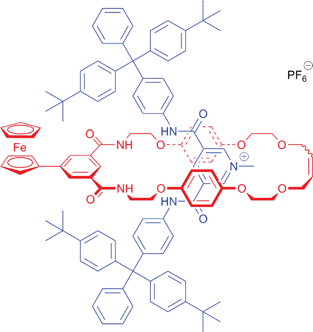 | ||
| Fig. 2 Target rotaxane 1+PF6−. | ||
Initial synthetic investigations were therefore undertaken to find appropriate conditions for the direct appendage of ferrocene to an isophthalamide motif by attempting the synthesis of acyclic model system 3 (Scheme 1). The attachment of ferrocene was achieved by following a literature procedure for the Suzuki coupling of ferroceneboronic acid with iodo-arene compounds,10 but using microwave irradiation for 30 min rather than refluxing for several days. Preparing iodo compound 2 and submitting it to these reaction conditions, followed by workup and column chromatography, allowed isolation of target compound 3 in a respectable yield of 58%. Model 3 was subsequently characterized by 1H and 13C NMR spectroscopy and high resolution mass spectrometry.
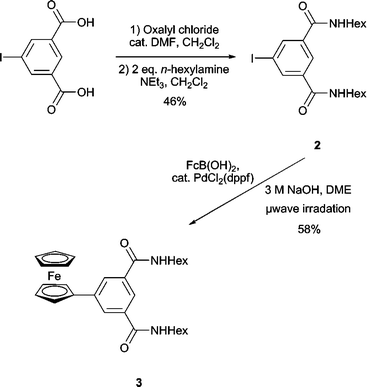 | ||
| Scheme 1 Synthesis of model 3. | ||
Having successfully demonstrated that it was possible to couple ferrocene to an isophthalamide motif, synthesis of target rotaxane 1+PF6− was carried out as shown in Schemes 2 and 3. Preparation of macrocycle precursor 5 was originally attempted by preparing compound 45c and exposing this to the same Suzuki conditions as for compound 2 (Route A in Scheme 2). Disappointingly, a repeatedly low yield (15–20%) for the formation of 5 was observed after workup and silica gel chromotography. In the belief that the presence of the terminal vinyl groups were interfering with the Suzuki coupling, an alternative route to 5 was pursued (Route B in Scheme 2). Pleasingly, the Suzuki coupling of compound 7 to form 8, occurred in a significantly higher yield of 55%. Removal of the benzyl protecting groups was achieved quantitatively by hydrogenation to yield 9 which was alkylated to produce the desired bis-vinyl appended precursor 5.
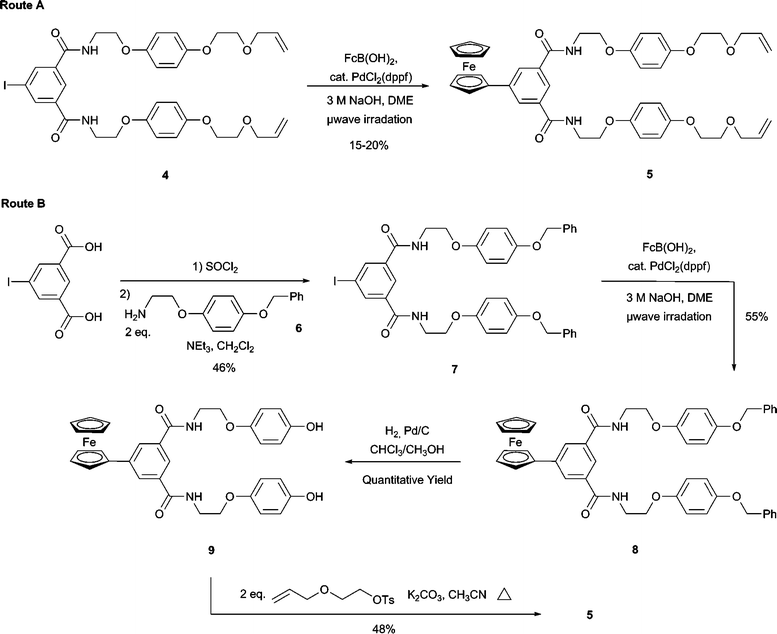 | ||
| Scheme 2 Synthetic routes to macrocyclic precursor 5. | ||
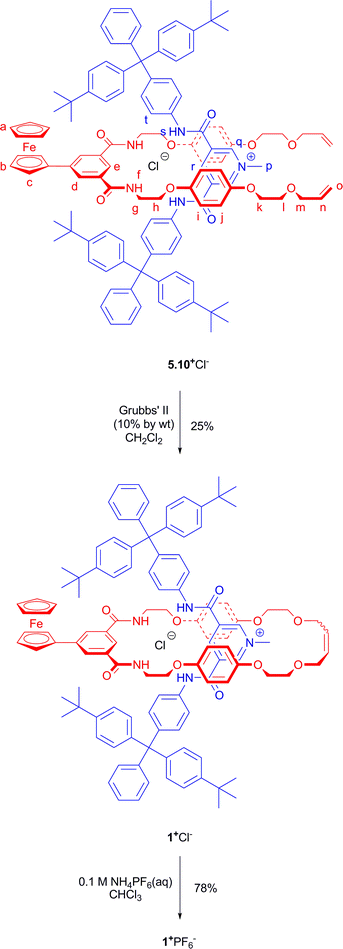
Rotaxane 1+Cl− was prepared by RCM reaction of equimolar amounts of 5 and methyl pyridinium chloride axle 10+Cl−4a in dichloromethane solution, in the presence of 10% (by wt) Grubbs' 2nd generation catalyst. The rotaxane was isolated in 25% yield after careful separation from other components in the reaction mixture by silica gel preparatory thin layer chromatography. Subsequent removal of the chloride ion template via anion exchange by washing with NH4PF6 furnished the desired rotaxane salt 1+PF6− (Scheme 3).
Both salts were characterized by 1H (1D and 2D) and 13C (and in the case of 1+PF6− by 19F and 31P) NMR spectroscopy and high resolution mass spectrometry.
Comparison of the 1H NMR spectra of the rotaxane 1+Cl− with macrocycle precursor 5 and axle 10+Cl− provides evidence of the interlocked nature of the rotaxane (Fig. 3). The success of the RCM reaction is indicated by the loss of peak o and the sharpening of multiplet n to a pseudo-singlet, these signals arising from the vinylic protons of the macrocycle precursor, with o lost as ethene, the by-product of the reaction. The spectra indicate three regions of interactions between the macrocyclic and axle components of the rotaxane. The first, the isophthalamide clefts, where strong upfield shifts in the axle signals r and s and downfield shifts in the macrocycle signals e and f, due to competitive hydrogen bond interactions between these protons and the chloride anion template, are observed. Second, the hydroquinone protons i and j have split and moved upfield, indicative of π–π stacking with the electron poor pyridinium unit. Finally, there is a large downfield shift in N-methyl peak p, indicating hydrogen bonding to the polyether oxygens of the macrocyclic component. Upon exchange to the hexafluorophosphate salt, there are upfield shifts of the isophthalamide cavity protons e, f, r and s, due to the removal of the chloride anion template. However, the hydroquinone protons i and j remain split and upfield compared to macrocycle precursor 5, indicating the π–π stacking between two components, and that the rotaxane species remains interlocked.
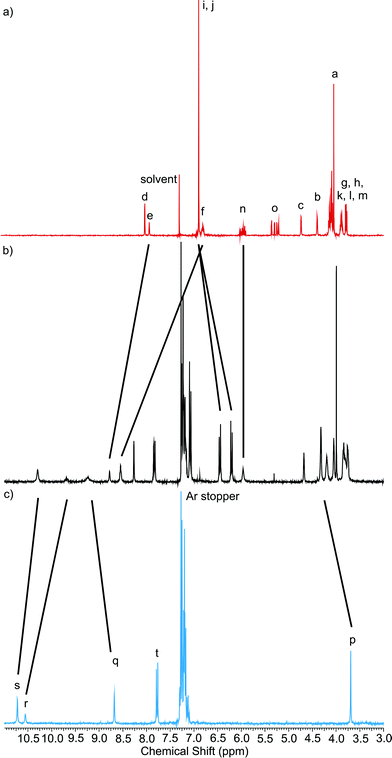 | ||
| Fig. 3 Partial 1H NMR spectra of a) macrocycle precursor 5, b) rotaxane 1+Cl− and c) axle 10+Cl− Solvent: CDCl3. See Scheme 3 for proton labels. | ||
The cationic fragment 1+ is to be found at m/z ∼ 1801.86 in high resolution electrospray mass spectra of both rotaxane salts. Conclusive evidence of the interlocked nature of the rotaxane chloride and hexafluorophophate salts was provided by the observation of numerous through space interactions between macrocycle and axle components in 2D ROESY 1H NMR spectra (see Fig. 4).
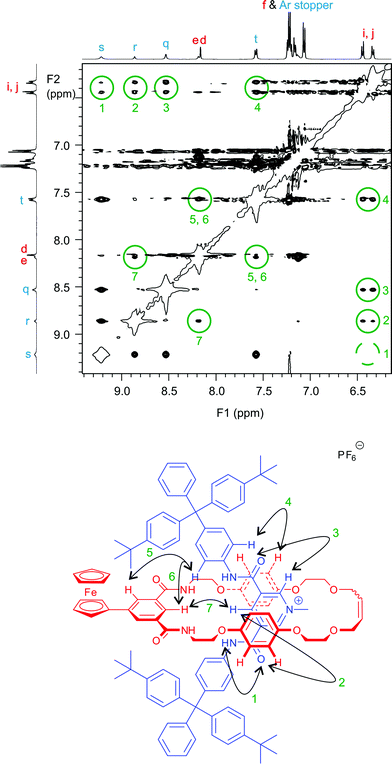 | ||
| Fig. 4 Section of 1H ROESY NMR spectrum of rotaxane 1+PF6− with through space intercomponent interactions highlighted. See Scheme 3 for proton labels. | ||
Anion recognition studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model (see Table 1).
:1 binding model (see Table 1).
With model 3, the anions are bound weakly in CD3CN and generally in the order of their basicity, i.e. benzoate > dihydrogen phosphate > hydrogen sulfate. Chloride is bound slightly more strongly than dihydrogen phosphate and hydrogen sulfate anions because of the size-fit complementarity with the isophthalamide cleft by the spherical monoatomic halide anion.12
An initial 1H NMR titration was undertaken with rotaxane 1+PF6− and TBACl in CD3CN. Exceptionally strong 1:1 binding was observed (K > 104 M−1). This is not unexpected: the rotaxane is positively charged, while the model is neutral. To allow for meaningful comparison of association constants of different anions with the rotaxane, the highly competitive solvent system 1:1 CDCl3:CD3OD was therefore used in subsequent titrations. There is a marked difference in selectivity of binding of the anion by rotaxane 1+PF6− in comparison to model 3. Most notably chloride - the least basic of the anions - is now bound the most strongly. Evidence of the origin of this is provided in the appearance of the titration curves (see Fig. 5). For chloride, the cleft proton of the pyridinium isophthalamide (proton r in Scheme 3) moves downfield upon addition of the anion, whereas for the oxoanions, this proton moves upfield. This is indicative of an alternative binding mode for these anions - presumably they are too large to penetrate into the cavity of the rotaxane and associate peripherally instead.
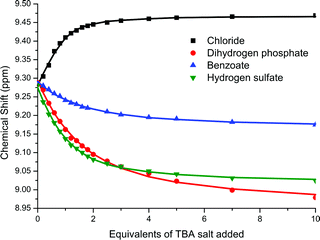
Model 3 exhibits a quasi-reversible oxidation for the Fc/Fc+ redox couple with E1/2 = +85 mV (compared to E1/2(ferrocene) = 0 V). Upon progressive addition of stoichiometric equivalents of anions cathodic shifts of the waves were observed: the maximum observed shifts in the redox wave being summarised in Table 2. These cathodic shifts can be attributed to the binding of the anion by the isophthalamide cleft protons, facilitating oxidation of ferrocene to ferrocenium. As chloride is added, a stepwise shifting of the quasi-reversible redox wave is observed, to a value of −20 mV upon addition of 10 equivalents of TBACl (see Fig. 6). In contrast, with dihydrogen phosphate and benzoate there is a loss of reversibility upon the addition of only small amounts of anion (1–2 equivalents). The disappearance of the reduction wave indicates either that the complexed anion-ferrocenium cation interaction is disfavouring reduction back to ferrocene or that an EC mechanism is in operation. Upon the addition of hydrogen sulfate the redox wave remains reversible throughout but only a modest cathodic shift is observed.
| Anion | ΔE1/2/mV |
|---|---|
| a Anions added as TBA salts. Electrolyte: 0.1 M TBAPF6 in CH3CN. Reference electrode: Ag/AgCl. Working electrode: Glassy Carbon. Auxiliary electrode: Platinum. 10 equivalents of anions added. Values reported to nearest 5 mV. T = 293 K. b Shift of oxidation peak, ΔEpa, due to loss of reversibility. | |
| Cl− | −20 |
| H2PO4− | −80b |
| BzO− | −10b |
| HSO4− | −10 |
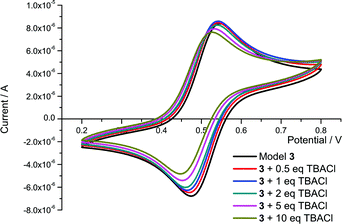 | ||
| Fig. 6 CVs of model 3 in 0.1 M TBAPF6/CH3CN upon the addition of aliquots of TBACl (Potential compared to Ag/AgCl reference). | ||
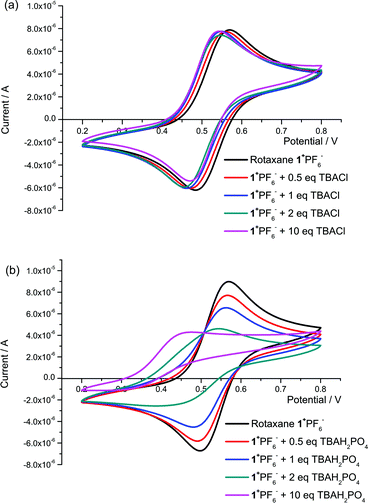
Rotaxane 1+PF6− also displays a quasi-reversible Fc/Fc+ redox couple, but with E1/2 = +115 mV (compared to E1/2(ferrocene) = 0 V). The more anodic potential for the rotaxane compared to acyclic model system 3 is expected as the rotaxane is positively charged which disfavours oxidation of the ferrocene moiety. Like the model, upon addition of chloride and hydrogen sulfate to samples of rotaxane, the redox couple remains quasi-reversible, while the addition of approximately 2 equivalents of dihydrogen phosphate or benzoate causes a loss of reversibility in the redox wave (see Fig. 7 for CVs of chloride and dihydrogen phosphate titrations). Not only is the magnitude of cathodic shift for chloride larger than any of the oxoanions at equimolar concentrations of rotaxane host and anion guest, but the cathodic shift of 20 mV is observed at 1 equivalent of added chloride anion with negligible further shift (< 5 mV) observed upon addition of further equivalents (see Table 3). These observations support the theory that only chloride is able to bind strongly within the interlocked cavity, with its presence being communicated to ferrocene presumably via a through-bond mechanism.13 With the oxoanions, there is a continued cathodic shift in the redox wave upon the addition of further equivalents of anion.§ Taking into account the 1H NMR anion binding results, which suggest the oxoanion association occurs on the periphery of the rotaxane structure, it is hypothesised that a through-space communication mechanism is in operation between the rotaxane's ferrocene redox centre and the oxoanion.13
| Anion | ΔE1/2/mV | |
|---|---|---|
| After 1 eq. of anion | After 10 eq. of anion | |
| a Anions added as TBA salts. Electrolyte: 0.1 M TBAPF6 in CH3CN. Reference electrode: Ag/AgCl. Working electrode: Glassy Carbon. Auxiliary electrode: Platinum. Values reported to nearest 5 mV. T = 293 K. b Shift of oxidation peak, ΔEpa, due to loss of reversibility. | ||
| Cl− | −20 | −20 |
| H2PO4− | −10 | −100b |
| BzO− | −15 | −25b |
| HSO4− | −15 | −40 |
Conclusions
A redox-active ferrocene functionalized rotaxane host system has been prepared by chloride anion templation. Upon exchange of the chloride anion template for the non-coordinating hexafluorophosphate anion 1H NMR anion titration investigations reveal that chloride is bound selectively over more basic oxoanions by the rotaxane: this is due to a complementary size match of the interlocked cavity for the monoatomic chloride anion, whereas oxoanions are too large to penetrate the cavity and associate peripherally. It has also been demonstrated that the rotaxane displays electrochemical recognition for chloride as evidenced by a maximum cathodic shift response of the Fc/Fc+ redox couple being observed at equimolar concentrations of rotaxane host and halide anion guest, whereas excess equivalents are required for oxoanions. The incorporation of redox-active groups into interlocked host design is continuing in our laboratories.Experimental
General
Commercially available solvents and chemicals were used without further purification unless stated. Where dry solvents were used, they were degassed with nitrogen, dried by passing through a MBraun MPSP-800 column and then used immediately. Deionised water was used in all cases. Triethylamine was distilled from and stored over potassium hydroxide. Thionyl chloride was distilled from triphenyl phosphate. Grubbs’ 2nd generation catalyst was stored in a desiccator. Microwave reactions were carried out using a Biotage Initiator 2.0 microwaveNMR spectra were recorded on Varian Mercury 300, Varian Unity Plus 500 and Bruker AVII 500 (with 13C Cryoprobe) spectrometers. Electrospray mass spectra were carried out on Micromass LCT and Bruker micrOTOF spectrometers. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Synthesis
5-Iodoisophthalic acid,14 iodo-arene precursor 4,4c amine 6,15 2-(allyloxy)ethyl 4-methylbenzenesulfonate16 and chloride axle 10+Cl−4a were prepared by literature procedure.:1 to 1:1) to yield iodo-arene 2 as a white solid (1.44 g, 46%). Mp = 100 °C; δH(300 MHz, CDCl3) 8.15 (2H, s, isophthalamide ArH4 & H6) 8.06 (1H, s, isophthalamide ArH2), 6.42 (2H, t, 3J = 5.3 Hz, NH), 3.40–3.46 (4H, m, NHCH2CH2), 1.56–1.66 (4H, app quartet, NHCH2CH2), 1.29–1.42 (12H, m, 3 × CH2), 0.88–0.92 (6H, m, CH3); δC(75.5 MHz, CDCl3) 165.5, 138.5, 136.7, 124.4, 94.3, 40.4, 31.5, 29.4, 26.7, 22.5, 14.0; m/z (ES) 459.1517 ([M + H]+, C20H32IN2O2 requires 459.1509), 459 (98%, [M + H]+), 476 (12, [M + NH4]+), 481 (98, [M + Na]+), 491 (65, [M + CH3OH + H]+), 497 (44, [M + K]+), 513 (100, [M + CH3OH + Na]+).
:0 to 2:1) to yield compound 3 as a dark orange solid (303 mg, 59%). Mp = 68–70 °C; δH(300 MHz, CDCl3) 7.97 (2H, d, 4J = 1.5 Hz, isophthalamide ArH4 & H6), 7.87 (1H, t, 4J = 1.5 Hz, isophthalamide ArH2), 6.32 (2H, t, 3J = 5.1 Hz, NH), 4.72 (2H, t, 3J = 1.8 Hz, Fc CpH), 4.38 (2H, t, 3J = 1.8 Hz, Fc CpH), 4.05 (5H, s, Fc Cp'H), 3.45–3.52 (4H, app quartet, NHCH2CH2), 1.60–1.70 (4H, m, NHCH2CH2), 1.31–1.46 (12H, m, 3 × CH2), 0.89–0.93 (6H, m, CH3); δC(75.5 MHz, CDCl3) 167.0, 141.2, 135.1, 127.1, 121.9, 83.1, 69.7, 69.6, 66.6, 40.3, 31.5, 29.5, 26.7, 22.5, 14.0; m/z (ES) 517.2513 ([M + H]+, C30H41FeN2O2 requires 517.2517), 517 (100%, [M + H]+), 539 (19, [M + Na]+), 549 (31, [M + CH3OH + H]+), 571 (14, [M + CH3OH + Na]+).
Route A. Ferroceneboronic acid (230 mg, 1.00 mmol) was added to DME (9 mL) in a 20 mL microwave vial (containing stirring bar). To this was added iodo-arene compound 4 (731 mg, 1.00 mmol), then 3 M NaOH(aq) solution (1 mL). This mixture was then degassed thoroughly for 20 min with nitrogen. PdCl2(dppf) (7 mg, 0.01 mmol) was then added, the microwave vial capped, and the vial subjected to microwave irradiation at 150 °C for 30 min. The reaction mixture was then diluted with CHCl3 (100 mL) and H2O (25 mL). The organic layer was separated, washed with H2O (3 × 50 mL) and dried over MgSO4, the solvent removed in vacuo and then purified by silica gel column chromatography (hexane–EtOAc 1
:1 to 1:2) to yield 5 as an orange oil (156 mg, 20%).
Route B. Diphenol compound 9 (250 mg, 0.40 mmol), 2-(allyloxy)ethyl 4-methylbenzenesulfonate (207 mg, 0.81 mmol) and K2CO3 (123 mg, 0.89 mmol) were suspended in dry CH3CN (15 mL) and heated under a nitrogen atmosphere for 60 h. The reaction was cooled to room temperature, filtered and the solvent removed in vacuo. The residue was dissolved in CH2Cl2, filtered and then dried (MgSO4), the solvent removed in vacuo, and the product isolated after silica gel column chromatography (CH2Cl2–CH3OH 98
:2) as an orange oil (151 mg, 48%).
δ
H(300 MHz, CDCl3) 7.99 (2H, d, 4J = 1.5 Hz, isophthalamide ArH4 & H6), 7.90 (1H, t, 4J = 1.5 Hz, isophthalamide ArH2), 6.86 (8H, app s, hydroquinone ArH), 6.78 (2H, t, 3J = 5.9 Hz, NH), 5.88–6.01 (2H, m, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.19–5.35 (4H, m, CHCH2), 4.72 (2H, app s, Fc CpH), 4.39 (2H, app s, Fc CpH), 4.07–4.15 (12H, m, 3 × CH2), 4.04 (5H, s, Fc Cp'H), 3.88 (4H, app quartet, NHCH2), 3.78 (4H, m, CH2); δC(75 MHz, CDCl3) 167.1, 153.2, 152.6, 141.3, 134.7, 134.5, 127.4, 122.1, 117.4, 115.6, 115.3, 82.9, 72.3, 69.7, 68.5, 68.0, 67.1, 66.7, 66.6, 39.8; m/z (ES) 811.2652 ([M + Na]+, C44H48FeN2NaO8 requires 811.2653), 789 (3%, [M + H]+), 806 (100, [M + NH4]+), 811 (67, [M + Na]+).
CH2), 5.19–5.35 (4H, m, CHCH2), 4.72 (2H, app s, Fc CpH), 4.39 (2H, app s, Fc CpH), 4.07–4.15 (12H, m, 3 × CH2), 4.04 (5H, s, Fc Cp'H), 3.88 (4H, app quartet, NHCH2), 3.78 (4H, m, CH2); δC(75 MHz, CDCl3) 167.1, 153.2, 152.6, 141.3, 134.7, 134.5, 127.4, 122.1, 117.4, 115.6, 115.3, 82.9, 72.3, 69.7, 68.5, 68.0, 67.1, 66.7, 66.6, 39.8; m/z (ES) 811.2652 ([M + Na]+, C44H48FeN2NaO8 requires 811.2653), 789 (3%, [M + H]+), 806 (100, [M + NH4]+), 811 (67, [M + Na]+).
:2), and then the resulting solid washed with EtOAc to give 7 as a white solid (1.83 g, 46%). Mp = 160 °C; δH(300 MHz, CDCl3) 8.22 (2H, d, 4J = 1.5 Hz, isophthalamide ArH4 & H6), 8.11 (1H, t, 4J = 1.5 Hz, isophthalamide ArH2), 7.32–7.44 (10H, m, benzyl ArH), 6.83–6.93 (8H, m, hydroquinone ArH), 6.69 (2H, t, 3J = 5.6 Hz, NH), 5.02 (4H, s, CH2), 4.10 (4H, t, 3J = 5.0 Hz, OCH2), 3.82–3.87 (4H, app. quartet, NCH2); δC(75 MHz, d6-DMSO) 164.6, 152.6, 152.5, 138.1, 137.3, 136.3, 128.4, 127.7, 127.6, 126.0, 115.7, 115.4, 94.6, 69.2, 66.3 (1 peak missing CNH - coincidental with solvent); m/z (ES) 765.1427 ([M + Na]+, C38H35IN2NaO6 requires 765.1432), 801 (100%, [M + CH3CN + NH4]+ - only assignable peak observed in low resolution mass spectrum.
:1) to yield 8 as a foaming orange solid (303 mg, 59%). Mp: phase transitions at 68 °C and 120 °C; δH(300 MHz, CDCl3) 8.00 (2H, d, 4J = 1.5 Hz, isophthalamide ArH4 & H6), 7.90 (1H, t, 4J = 1.5 Hz, isophthalamide ArH2), 7.32–7.44 (10H, m, benzyl ArH), 6.86–6.94 (8H, m, hydroqunione ArH), 6.71–6.75 (2H, t, 3J = 5.7 Hz, NH), 5.02 (4H, s, CH2), 4.72–4.73 (2H, m, Fc CpH), 4.38–4.39 (2H, m, 2H, Fc CpH), 4.13–4.16 (4H, m, OCH2), 4.04 (5H, s, Fc Cp'H), 3.87–3.92 (4H, app. quartet, NCH2); δC(75 MHz, CDCl3) 167.1, 153.3, 152.7, 141.4, 137.1, 134.8, 128.5, 127.9, 127.4, 122.1, 115.9, 115.4, 82.9, 70.6, 69.7, 69.7 (sic), 67.2, 66.7, 39.8. (1 ArC peak missing); m/z (ES) 823.2436 ([M + Na]+, C48H44FeN2NaO6 requires 823.2442), 823 (7%, [M + Na]+), 859 (100, [M + CH3CN + Na]+).
:1, 20 mL) was added 10% Pd/C (10% by wt, 40 mg), and the reaction was stirred under an atmosphere of H2 for 16 h. The reaction mixture was filtered through Celite®, the solvent removed in vacuo, to leave the product as a foaming orange solid (310 mg, quant.). Mp > 148 °C (dec.); δH(300 MHz, d6-DMSO) 8.94 (2H, s, OH), 8.87 (2H, t, 3J = 5.6 Hz, NH), 8.20 (1H, t, 4J = 1.5 Hz, isophthalamide ArH2), 8.13 (2H, d, 4J = 1.5 Hz, isophthalamide ArH4 & H6), 6.65–6.82 (8H, m, hydroquinone ArH), 4.93 (2H, t, 3J = 1.9 Hz, Fc CpH), 4.43 (2H, t, 3J = 1.9 Hz, Fc CpH), 4.02–4.07 (9H, m, Fc Cp'H & OCH2), 3.60–3.66 (4H, app. quartet, NCH2); δC(75 MHz, d6-DMSO) 166.0, 151.4, 151.2, 139.9, 134.6, 126.7, 124.1, 115.8, 115.5, 83.4, 69.5, 69.4, 66.6, 66.6 (sic) (1 peak missing CNH - coincidental with solvent); m/z (ES) 643.1504 ([M + Na]+, C34H32FeN2NaO6 requires 643.1502), 643 (5%, [M + Na]+), 679 (100, [M + CH3CN + NH4]+). In addition, 619 (100%, [M − H]−), 655 (43, [M + Cl]−) observed in negative polarity mass spectrum.
:4, then EtOAc), rotaxane 1+Cl− was isolated as an orange solid (48 mg, 25%). Mp > 230 °C (dec.); δH(300 MHz, CDCl3): 10.30 (2H, s, py NH), 9.68 (1H, s, pyridinium ArH4), 9.25 (2H, s, pyridinium ArH2 & ArH6), 8.77 (1H, s, isophthalamide ArH2), 8.55 (2H, s, isophthalamide NH), 8.26 (2H, s, isophthalamide ArH4 & ArH6), 7.84 (4H, d, 3J = 8.8 Hz, stopper NHArH), 7.06–7.26 (30H, m, stopper ArH), 6.45 (4H, d, 3J = 8.9 Hz, hydroquinone ArH), 6.20 (4H, d, 3J = 8.9 Hz, hydroquinone ArH), 5.96 (2H, br s, CHCH), 4.68 (2H, app s, Fc CpH), 4.32 (5H, app s, Fc CpH & N+CH3), 4.19 (4H, br s, CH2), 4.05 (app s, 4H, CH2), 4.00 (5H, s, Fc Cp'H), 3.75–3.85 (12H, m, 3 × CH2), 1.32 (36H, s, (CH3)3); δC(125.8 MHz, CDCl3) 168.9, 158.1, 153.4, 151.9, 148.4, 146.9, 145.2, 144.8, 143.5, 140.4, 139.4, 134.8, 134.0, 133.4, 131.8, 131.1, 130.6, 129.9, 129.0, 127.4, 125.8, 124.3, 122.1, 120.4, 114.8, 114.5, 71.0, 69.3, 68.0, 66.1, 63.8, 48.8, 40.6, 34.3, 31.4 (in addition evidence of broad peaks attributed to the four CpC); m/z (ES) 1800.8585 ([M − Cl]+, C116H122FeN5O10 requires 1800.8541), 1801 (100%, [M − Cl]+).
CH), 4.73 (2H, s, Fc CpH), 4.37 (2H, s, Fc CpH), 4.31 (3H, s, N+CH3) 4.04–4.06 (9H, m, Fc Cp'H & CH2) 3.93 (4H, t, 3J = 4.4 Hz, CH2), 3.86 (4H, m, CH2), 3.81 (4H, m, CH2) 3.70–3.72 (4H, m, CH2), 1.31 (s, 36H, (CH3)3); δC(125.8 MHz, CDCl3) 167.0, 158.3, 152.8, 152.0, 148.6, 146.8, 145.3, 144.0, 143.4, 141.8, 141.3, 134.3, 134.3 (sic), 134.1, 132.0, 131.0, 130.6, 130.4, 128.5, 127.4, 125.9, 124.4, 121.6, 120.1, 115.5, 114.7, 83.2, 70.8, 69.7, 69.5, 69.3, 67.7, 66.9, 66.8, 63.9, 49.6, 39.7, 34.3, 31.4; δF(282.4 MHz, CDCl3) −69.9 (d, 1J = 714 Hz, PF6); δP(121.5 MHz, CDCl3) −143.9 (septet, 1J = 714 Hz, PF6−); m/z (ES) 1800.8605 ([M − PF6]+, C116H122FeN5O10 requires 1800.8541), 1801 (100%, [M − PF6]+).
Acknowledgements
We wish to thank the EPSRC for a DTA studentship (N. H. E.), Johnson-Matthey for the loan of palladium catalysts, and Dr J. J. Davis (University of Oxford) for use of electrochemical equipment.Notes and References
- For general reviews on rotaxanes and catenanes see: (a) T. Takata, N. Kihara and Y. Furusho, Adv. Poly. Sci., 2004, 171, 1 CAS; (b) J.-P. Collin, V. Hertz and J.-P. Sauvage, Top. Curr. Chem., 2005, 262, 29 CAS; (c) F. H. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982 CrossRef CAS; (d) G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782 CrossRef CAS; (e) M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657 CrossRef CAS; (f) K. E. Griffiths and J. F. Stoddart, Pure Appl. Chem., 2008, 80, 485 CrossRef CAS; (g) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974 CrossRef CAS; (h) J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329 RSC; (i) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530 RSC; (j) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024 CrossRef CAS; (k) For a comprehensive review on the nanotechnological application of interlocked molecules see: E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 Search PubMed.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415 RSC.
- Examples of cation-binding rotaxanes: (a) K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568 CrossRef CAS; (b) N. C. Chen, P. Y. Huang, C. C. Lau, Y. H. Liu, Y. Wang, S. M. Peng and S. H. Chiu, Chem. Commun., 2007, 4122 RSC; (c) S. S. Zhu, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713 CrossRef CAS; (d) S. S. Zhu and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 12568 CrossRef CAS.
- Examples of anion-binding rotaxanes: (a) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469 CrossRef CAS; (b) D. Curiel and P. D. Beer, Chem. Commun., 2005, 1909 RSC; (c) M. R. Sambrook, P. D. Beer, M. D. Lankshear, R. F. Ludlow and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 1529 RSC; (d) S. R. Bayly, T. M. Gray, M. J. Chmielewski, J. J. Davis and P. D. Beer, Chem. Commun., 2007, 2234 RSC; (e) L. M. Hancock and P. D. Beer, Chem.–Eur. J., 2009, 15, 42 CrossRef CAS; (f) L. Y. Zhao, J. J. Davis, K. M. Mullen, M. J. Chmielewski, R. M. J. Jacobs, A. Brown and P. D. Beer, Langmuir, 2009, 25, 2935 CrossRef CAS; (g) A. Brown, K. M. Mullen, J. Ryu, M. J. Chmielweski, S. M. Santos, V. Felix, A. L. Thompson, J. E. Warren, S. I. Pascu and P. D. Beer, J. Am. Chem. Soc., 2009, 131, 4937 CrossRef CAS; (h) K. M. Mullen, J. Mercurio, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2009, 48, 4781 CrossRef CAS; (i) A. J. McConnell, C. J. Serpell, A. L. Thompson, D. R. Allan and P. D. Beer, Chem.–Eur. J., 2010, 16, 1256 CrossRef CAS; (j) J. J. Gassensmith, S. Matthys, J. J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem.–Eur. J., 2010, 16, 2916 CrossRef.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC, Cambridge, 2006 Search PubMed.
- Illustrative examples of ferrocene-containing anion receptors: (a) P. D. Beer, Z. Chen, A. J. Goulden, A. Graydon, S. E. Stokes and T. Wear, J. Chem. Soc., Chem. Commun., 1993, 1834 RSC; (b) P. D. Beer, A. R. Graydon, A. O. M. Johnson and D. K. Smith, Inorg. Chem., 1997, 36, 2112 CrossRef CAS; (c) G. Denuault, P A. Gale, M. B. Hursthouse, M. E. Light and C. N. Warriner, New J. Chem., 2002, 26, 811 RSC; (d) Y. Willener, K. A. Joly, C. J. Moody and J. H. R. Tucker, J. Org. Chem., 2008, 73, 1225 CrossRef CAS; (e) F. Zapata, A. Caballero, A. Tarraga and P. Molina, J. Org. Chem., 2010, 75, 162 CrossRef CAS. For reviews see: (f) P. D. Beer and S. R. Bayly, Top. Curr. Chem., 2005, 255, 125 CAS; (g) S. R. Bayly, P. D. Beer and G. Z. Chen, in Ferrocenes: Ligands, Materials and Biomolecules, ed. Petr Štěpnička, Wiley, UK, 2008, ch. 8, pp 281–318 Search PubMed.
- (a) N. Kihara, M. Hashimoto and T. Takata, Org. Lett., 2004, 6, 1693 CrossRef CAS; (b) R. Isnin and A. E. Kaifer, J. Am. Chem. Soc., 1991, 113, 8188 CrossRef; (c) A. C. Benniston and A. Harriman, Angew. Chem., Int. Ed. Engl., 1993, 32, 1459 CrossRef; (d) A. C. Benniston and A. Harriman, J. Am. Chem. Soc., 1995, 117, 5275 CrossRef CAS.
- (a) I. Willner, V. Pardo-Yissar, E. Katz and K. T. Ranjit, J. Electroanal. Chem., 2001, 497, 172 CrossRef CAS; (b) A. Mateo-Alonso, C. Ehli, G. M. A. Rahman, D. M. Guldi, G. Fioravanti, M. Maraccio, F. Paolucci and M. Prato, Angew. Chem., Int. Ed., 2007, 46, 3521 CrossRef CAS; (c) Y. Suzuki, E. Chihara, A. Takagi and K. Osakada, Dalton Trans., 2009, 9881 RSC.
- During the course of this work another group published an article on the synthesis and electrochemical anion recognition of a set of simple acyclic 5-ferrocenylisophthalamides: W. Liu, H. Zheng, L. Du, B. Chen and M. Song, Z. Anorg. Allg. Chem., 2010, 636, 236 Search PubMed.
- R. Knapp and M. Rehahn, J. Organomet. Chem., 1993, 452, 235 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- K. Kavallieratos, C. M. Bertao and R. H. Crabtree, J. Org. Chem., 1999, 64, 1675 CrossRef CAS.
- P. D. Beer, P. A. Gale and G. Z. Chen, J. Chem. Soc., Dalton Trans., 1999, 1897 RSC.
- A. Kraft, Liebigs Ann./Recl., 1997, 1463 Search PubMed.
- C. Goldenberg, R. Wandestrick, F. Binon and R. C Chorlier, Chim. Ther., 1973, 8, 259 CAS.
- H. D. Maynard and R. H. Grubbs, Macromolecules, 1999, 32, 6917 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral characterisation of novel compounds; protocols for 1H NMR and electrochemical titrations, plus titration data. See DOI: 10.1039/c0ob00458h |
| ‡ While perhaps being more appropriate for comparison to rotaxane 1+PF6− than acyclic model 3, the macrocyclic component of the rotaxane proved very difficult to isolate pure in sufficient quantities for anion recognition studies. In addition, it was also found to have low solubility in acetonitrile, the solvent used in the electrochemistry investigations. |
| § Attempts at electrochemical anion recognition competition experiments (e.g. addition of chloride to rotaxane 1+PF6− in the presence of excess equivalents of oxoanions) were undertaken but proved inconclusive due to irreversible electrochemical behaviour. |
| This journal is © The Royal Society of Chemistry 2011 |