A combined mechanistic and computational study of the gold(I)-catalyzed formation of substituted indenes†
Pierrick
Nun
a,
Sylvain
Gaillard
a,
Albert
Poater
b,
Luigi
Cavallo
c and
Steven P.
Nolan
*a
aEasStCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, UK KY16 9ST. E-mail: snolan@st-andrews.ac.uk
bCatalan Institute for Water Research (ICRA), H2O Building, Scientific and Technological Park of the University of Girona, Emili Grahit 101, E-17003 Girona, Spain
cDipartimento di Chimica, Università di Salerno, Via ponte don Melillo, 84084, Fisciano, Italy
First published on 20th October 2010
Abstract
Substituted indenes can be prepared after a sequence [1,3] O-acyl shift-hydroarylation-[1,3] O-acyl shift. Each step is catalyzed by a cationic NHC-Gold(I) species generated in situ after reaction between [(IPr)AuOH] and HBF4·OEt2. This interesting silver-free way is fully supported by a computational study justifying the formation of each intermediate.
Introduction
The use of gold as a molecular assembly strategy is growing rapidly in interest as attested by the expanding related literature.1 Among the most prevalent transformations examined, those involving alkynes,2 with specific examples making use of propargylic esters, are most common.3Propargylic acetates I have already been shown to react in the presence of [(IPr)AuCl]/AgBF4 (IPr = 1,3-bis(diisopropyl)phenylimidazol-2-ylidene) through a tandem [3,3] rearrangement-intramolecular hydroarylation to give either indenes III or IV.4 The same reaction in presence of water leads to enone V (Scheme 1).5 We recently described the synthesis and characterization of a NHC-gold(I) hydroxide complex.6 This compound displays a strong Brønsted basic character and can react with acids to generate a cationic gold(I) species.7 This catalytic system is user-friendly and circumvents the need of silver salts in association with gold to generate the putative [NHC–Au]+ species. In the present study, this simple activation protocol ([(IPr)AuOH] + HBF4·OEt2) was examined in a skeleton rearrangement reaction involving propargylic acetates. (Scheme 2).8
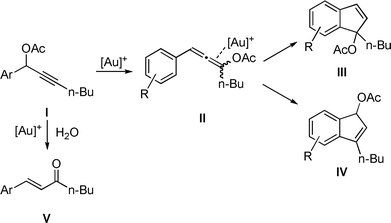 | ||
| Scheme 1 Transformations of propargylic acetates. | ||
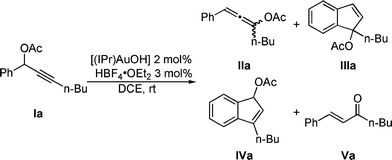
Results and discussion
Propargylic acetate Ia was fully converted after 5 min at RT in the presence of [(IPr)Au]+ formed in situ using the acid activation method described above.As previously observed,4,9 products IIa and IIIa were formed along with trace amounts (<5%) of enone Va.5 The reaction mixture was analyzed after 5 h and surprisingly another compound identified as indene IVa appeared in a IIIa/IVa ratio 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13. After 24 h this ratio evolved to 70:30 (IIIa/IVa). These results led us to reconsider the previously proposed mechanism for this transformation. Indeed, indene IVa appears to be a rearranged product of IIIa and not a product obtained directly from allene IIa. The influence of temperature on conversion of Ia into IVa was next investigated. Complete conversion at 25 °C, 35 °C and 45 °C was reached after 30 h, 14 h and 4 h, respectively. Monitoring the progress of the reaction by 1H NMR spectroscopy clearly illustrates the progress of the transformation (Fig. 1).
:13. After 24 h this ratio evolved to 70:30 (IIIa/IVa). These results led us to reconsider the previously proposed mechanism for this transformation. Indeed, indene IVa appears to be a rearranged product of IIIa and not a product obtained directly from allene IIa. The influence of temperature on conversion of Ia into IVa was next investigated. Complete conversion at 25 °C, 35 °C and 45 °C was reached after 30 h, 14 h and 4 h, respectively. Monitoring the progress of the reaction by 1H NMR spectroscopy clearly illustrates the progress of the transformation (Fig. 1).
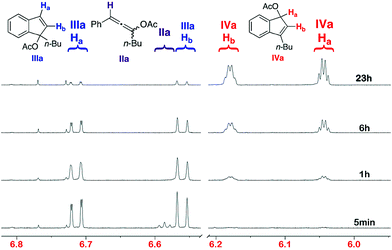 | ||
| Fig. 1 1H NMR of the reaction mixture at room temperature. | ||
After 5 min., complete conversion of Ia into the allene IIa (triplet at 6.58 ppm), indene IIIa (doublets at 6.70 and 6.56 ppm) and enone Va (doublet at 6.75 ppm) was observed. After 1 h, allene IIa was totally converted into indene IIIa. After 6 h and 23 h, a constant increase of the proportion of IVa linked to a decrease of IIIa was noticed. No intermediate between these two compounds was observed, suggesting that a direct [1,3]-shift of the acetate group is involved in the transformation.
To further support the proposed mechanism, a computational analysis was performed.10Fig. 2 illustrates all the intermediates found for the Ia → IIa → IIIa → IVaisomerization.
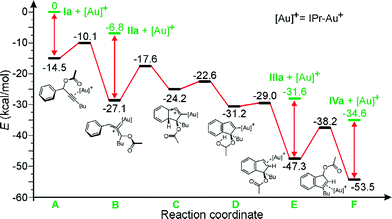 | ||
| Fig. 2 Energy profile for the Ia → IIa → IIIa → IVa conversion. | ||
After coordination of Ia to the Au center, the proposed [1,3] shift of the acetyl group requires to overcome of a barrier of only 4.4 kcal mol−1, evolving the system into intermediate B. Intermediate B has a strong Au-carbenoid character, as indicated by the strongly bent allene moiety (the C–C–C allene angle in B is 127.8°). Dissociation of IIa from B requires 20.3 kcal mol−1. The hydroarylation B–C step is endothermic by 9.5 kcal mol−1, and leads to intermediate C, which proceeds with a low barrier to intermediate D, recovering the aromaticity of the arene ring of the substrate. The hydrogen of the hydroxyl group of D then migrates to the C atom σ-bonded to the metal in D. The D–E step is nearly thermoneutral, and E is the first intermediate that is remarkably more stable than B. Dissociation of IIIa from E costs 15.7 kcal mol−1. Finally, a second [1,3] shift migration of the acetyl group of E occurs into the final position in F with a barrier of 9.1 kcal mol−1 only. Dissociation of the most stable product IVa from F is endothermic by 18.9 kcal mol−1. In short, the proposed reaction pathway is fully supported by the theoretical analysis. All barriers connecting Ia to IVa are lower than 10 kcal mol−1, and product IVa is the most stable product. Further, the mechanistic sequence Ia → IIa → IIIa → IVa is in agreement with the NMR analysis of Fig. 1. On the other hand, direct hydroarylation from A, i.e. without the initial [1,3] shift of the acetyl group, has the high barrier of 17.8 kcal mol−1. Similarly, the direct H transfer from B, i.e. before the hydroarylation, has a barrier of 44.0 kcal mol−1. This indicates that the sequence of steps illustrated in Fig. 2 is the only viable one. Finally, the barrier of 32.4 kcal mol−1 for the IIIa → IVaisomerization in the absence of the Au-catalyst, excludes the possibility of this transformation proceeding in the absence of gold.
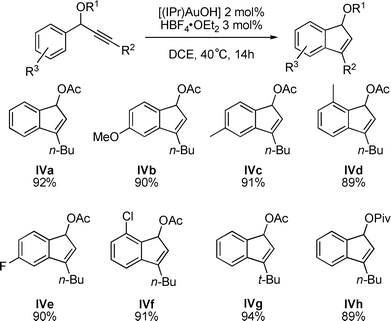
This reaction was extended to other propargylic acetates leading to a variety of indenes (Fig. 3).11 Various substitutions, electron withdrawing or donating, on the aromatic ring as well as the effect of a more hindered O-Acyl group (IVh) instead of the acetate were studied.
 | ||
| Fig. 3 Gold-catalyzed synthesis of indenes. | ||
In all cases, the sequential formation of allene II, kinetic indene III and then thermodynamic indene IV was observed by 1H NMR spectroscopy. The presence of electron donating (IVa–d) as well as halogenes (IVe–f) on the aromatic ring in ortho and in para positions showed no effect on the hydroarylation step leading to the corresponding indenes in high yields (>89%). The reaction is also quite efficient if the n-butyl chain is replaced by a more sterically demanding t-butyl (IVg). We also investigated the reactivity of a pivaloyl substituent as a migrating group, with no notable effect of the more hindered substitution (IVh).
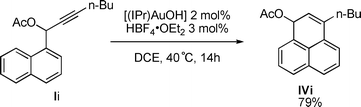
Interestingly, product IVi can also be prepared from propargylic acetate Ii through the same sequence after formation of a 6-membered ring preceding the [1,3] O-acyl shift but without any indene formation (eqn (1)).
 | (1) |
To support our mechanistic hypothesis, isolated allene IIa and indene IIIa were reacted under catalytic conditions. As expected, in the presence of our catalytic system, IIa or IIIa led quantitatively to indene IVa. Simple heating of these substrates with or without HBF4·OEt2 (3 mol%) did not yield IVa, excluding the simple thermal or acid-catalyzed rearrangement reaction pathway.
Conclusion
In conclusion, the pre-catalyst [(IPr)AuOH] is shown to be useful in the preparation of substituted indenes in an entirely silver-free manner viaactivation with HBF4·OEt2 as a Brønsted acid to generate the active species in situ. The use of this new catalytic system permitted the examination of individual steps involved in the transformation and to correct the previously proposed mechanism. The proposed steps are fully supported by a computational study. The transformation involves firstly a [1,3] O-acyl shift to form an allene, then hydroarylation to produce a kinetically favored indene and finally another [1,3] O-acyl shift generates a more stable substituted indene.Experimental section
All reagents and solvents were used as purchased. NMR spectra were recorded on a 400 MHz Varian Gemini spectrometer. High Resolution Mass Spectrometry analyses were performed by St Andrews analytical services.Complex [(IPr)AuOH]6 and propargylic acetates4,5,12 were prepared according to literature procedures. For computational data see supporting information.†
Computational details
All the DFT static calculations were performed at the GGA level with the Gaussian03 set of programs,13 using the BP86 functional of Becke and Perdew.14 The electronic configuration of the molecular systems was described with the standard split-valence basis set with a polarization function of Ahlrichs and co-workers for H, C, N, and O (SVP keyword in gaussian).15 For Au we used the small-core, quasi-relativistic Stuttgart/Dresden effective core potential, with an associated valence basis set contracted (standard SDD keywords in gaussian03).16 The geometry optimizations were performed without symmetry constraints, and the characterization of the located stationary points was performed by analytical frequency calculations. The energies discussed throughout the text contain zero point energy (ZPE) corrections. Solvent effects including contributions of non electrostatic terms have been estimated in single point calculations on the gas phase optimized structures, based on the polarizable continuous solvation model PCM using dichloroethane as solvent.17General procedure for the synthesis of indenes
In a flame-dried Schlenk, 2.4 mg of IPrAuOH (6.10−3 mmol, 2 mol%) is diluted in 8 ml of 1,2-dichloroethane, then 84 μl of a 1% solution of HBF4·OEt2 in dichloroethane (9.10−3 mmol, 3 mol%) is added. Mixture is stirred at room temperature for 5 min, then a solution of propargylic acetate (0.3 mmol, 1 eq) in 2 ml of dichloroethane is added. Reaction is heated overnight at 40 °C. The solvent is evaporated and the crude product diluted in pentane and filtered over a plug of silica to remove catalyst or purified on column chromatography if needed.Acknowledgements
The ERC (Advanced Investigator Award to SPN) and the EPSRC are gratefully acknowledged for support of this work. Umicore AG are thanked for their generous gifts of materials. LC thanks the HPC team of Enea (www.enea.it) for using the ENEA-GRID and the HPC facilities CRESCO (www.cresco.enea.it) in Portici, Italy. AP thanks the Spanish MICINN for a Ramón y Cajal contract. SPN is a Royal Society-Wolfson Research Merit Award holder.Notes and references
- For selected recent reviews on gold-catalyzed reactions, see: (a) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–322 RSC; (b) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS; (c) P. Belmont and E. Parker, Eur. J. Org. Chem., 2009, 6075–6089 CrossRef CAS; (d) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef; (e) D. J. Gorin, F. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS; (f) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS; (g) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef; (h) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef; (i) E. Jiménez-Nunez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef CAS; (j) R. Skouta and C.-J. Li, Tetrahedron, 2008, 64, 4917–4938 CrossRef CAS; (k) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC; (l) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS.
- (a) A. Martínez, P. García-García, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Angew. Chem., Int. Ed., 2010, 49, 4633–4637 CrossRef CAS; (b) E. Jiménez-Nunez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC.
- (a) G. Li, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 3740–3741 CrossRef CAS; (b) A. Correa, N. Marion, L. Fensterbank, M. Malacria, S. P. Nolana and L. Cavallo, Angew. Chem., Int. Ed., 2008, 47, 718–721 CrossRef CAS; (c) N. Marion, G. Lemière, A. Correa, C. Costabile, R. S. Ramon, X. Moreau, P. De Frémont, R. Dahmane, A. Hours, D. Lesage, J.-C. Tabet, J.-P. Goddard, V. Gandon, L. Cavallo, L. Fensterbank, M. Malacria and S. P. Nolan, Chem.–Eur. J., 2009, 15, 3243–3260 CrossRef CAS; (d) D. Wang, X. Ye and X. Shi, Org. Lett., 2010, 12, 2088–2091 CrossRef CAS; (e) D. Garayalde, E. Gomez-Bengoa, X. Huang, A. Goeke and C. Nevado, J. Am. Chem. Soc., 2010, 132, 4720–4730 CrossRef CAS.
- N. Marion, S. Díez-González, P. De Frémont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647–3650 CrossRef CAS.
- N. Marion, P. Carlqvist, R. Gealageas, P. De Frémont, F. Maseras and S. P. Nolan, Chem.–Eur. J., 2007, 13, 6437–6451 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS.
- The amount of acid used (3 mol%) was selected for ease of manipulation. Background reactions conducted with acid only did not lead to product formation and permitted recovery of starting material.
- N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750–2752 CrossRef CAS.
- The BP86 DFT calculations were performed with the Gaussian03 package using a SVP basis set on main group atoms, and the SDD-ECP basis set on Ru. Solvent effects, dichloroethane, were included with the PCM approach. Further details in the Supporting Information.
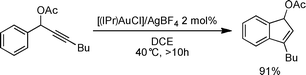
- The reaction below conducted under identical catalytic conditions but with the use of a Au–Cl precursor activated by a silver salt yields comparable results (thermodynamic indene) but with apparent slower kinetics as under optimised conditions this transformation mediated by [(IPr)AuOH] and acid reaches completion after 4 h
. - M. Yu, G. Zhang and L. Zhang, Org. Lett., 2007, 9, 2147–2150 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, J., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, Ö. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian03, Revision E.01; Gaussian Inc.: Wallingford CT, 2004 Search PubMed.
- (a) A. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef; (c) J. P Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406–7406 CrossRef.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- (a) U. Haeusermann, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1993, 78, 1211–1224; (b) W. Kuechle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef; (c) T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational data and NMR spectra. See DOI: 10.1039/c0ob00758g |
| This journal is © The Royal Society of Chemistry 2011 |