Amino acids attached to 2′-amino-LNA: synthesis and excellent duplex stability†
Marie W.
Johannsen
,
Lia
Crispino
,
Michael C.
Wamberg
,
Neerja
Kalra
and
Jesper
Wengel
*
Nucleic Acid Center, Department of Physics and Chemistry, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark.. E-mail: jwe@ifk.sdu.dk; Fax: +45 6550 4385; Tel: +45 6550 2510
First published on 3rd November 2010
Abstract
The synthesis of 2′-amino-LNA (the 2′-amino derivative of locked nucleic acid) has opened up a number of exciting possibilities with respect to modified nucleic acids. While maintaining the excellent duplex stability inferred by LNA-type oligonucleotides, the nitrogen in the 2′-position of 2′-amino-LNA monomers provides an excellent handle for functionalisation. Herein, the synthesis of amino acid functionalised 2′-amino-LNA derivatives is described. Following ON synthesis, a glycyl unit attached to the N2′-position of 2′-amino-LNA monomers was further acylated with a variety of amino acids. On binding to DNA/RNA complements, the modified ONs induce a marked increase in thermal stability, which is particularly apparent in a buffer system with a low salt concentration. The increase in thermal stability is thought to be caused, at least in part, by decreased electrostatic repulsion between the negatively charged phosphate backbones when positively charged amino acid residues are appended. Upon incorporation of more than one 2′-amino-LNA modification, the effects are found to be nearly additive. For comparison, 2′-amino-LNA derivatives modified with uncharged groups have been synthesised and their effect on duplex thermal stability likewise investigated.
Introduction
In recent years, an abundance of modified oligonucleotides (ONs) have been presented. These contain modifications of the sugar ring and its substituents,1 the nucleobases2,3 or the backbone.4 Previous research in our group has focused on locked nucleic acid (LNA, Fig. 1). LNA is a nucleotide analogue where the sugar ring is constrained as part of a bicyclic system.5,6 This locks the furanose in the C3′-endo conformation, thus mimicking the conformation found in RNA. ONs modified with LNA nucleotides have a high affinity for both complementary DNA and RNA strands, as evidenced by increased duplex thermal denaturation temperatures. 2′-Substituted analogues of LNA have been developed, including 2′-thio-LNA and 2′-amino-LNA (Fig. 1).7,8 Unlike 2′-amino-DNA, which destabilises both DNA and RNA type helix formation,9,102′-amino-LNA has a higher affinity for complementary DNA/RNA strands than unmodified ONs, though 2′-amino-LNA is slightly less stabilising than LNA.8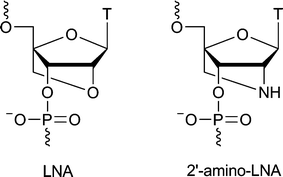 | ||
| Fig. 1 Structures of LNA and 2′-amino LNA thymine monomers. | ||
Pharmacokinetic challenges, including delivery, potentially limit realisation of the many possible pharmacological uses of ONs.11–13 Increasing the structural diversity of ONs is one possible way of achieving ONs with drug-like characteristics, and modification of the nucleotide building blocks may increase the stability towards nuclease degradation, affinity for complementary strands, potency and biodistribution of therapeutic ONs.4,14–16
A large area of research is the conjugation of various functional groups and molecules to ONs.17,18 Recently, many conjugations have been performed using Huisgen–Sharpless–Meldal reactions (more commonly known as “click”-chemistry)19 and other bio-orthogonal chemical reactions. Conjugations can be carried out both during and after ON synthesis, and may take place at the 5′-end, 3′-end or internally in the ON.15,17,18 A major obstacle to delivery of ONs is cell penetration. Negatively charged proteoglycans on the cell membrane repel the polyanionic ONs, and the hydrophobic nature of the cell membrane hampers entry of hydrophilic ONs. Proposed solutions for this problem include neutral backbone ONs20 and conjugation to various delivery agents such as cholesterol21,22 or cell penetrating peptides (CPPs). CPPs are short sequences of amino acids which, when conjugated to biopharmaceuticals, have been found to increase cellular uptake and biological activity of their cargo.23–25
It is intriguing that ONs with several positive amino acid residues (e.g. lysines) appended can have a neutral or positive net charge while maintaining water solubility. Several studies on cationic ONs, both with positively charged backbones and with positively charged groups attached have been reported.15,26–28 Several ONs modified with cationic groups in the 2′-position exhibit efficient binding affinity to target RNA, enhanced chemical stability and increased nuclease resistance.29
In this paper, we present various structures based on a 2′-amino-LNA/DNA mixmer scaffold. We have previously used LNA to counter the destabilising effect of the incorporation of 2′-amino-DNA monomers with and without modifications of the 2′-amino group.10,30 The research described here instead focuses on conjugation of amino acids and peptides to the thermally stabilising 2′-amino-LNA monomer. The amino group of 2′-amino-LNA nucleotides is an excellent site for functionalisation of ONs.31–37Conjugation to a 2′-amino group is potentially more selective and easy than conjugation to a 2′-hydroxy group of RNA, and the nitrogen atom furthermore allows functionalisation even when the 2′-nitrogen atom is partaking in the bicyclic ring skeleton of an LNA nucleotide.
We report here the design and synthesis of various 2′-amino-LNA phosphoramidites functionalised at the N2′ position by the amino acid glycine (“gly”), acetic acid (“acetyl”), and palmitic acid (“C16”), and incorporation of these into ONs. Conjugation of palmitic acid to ONs has previously been investigated with the intention of facilitating cellular uptake,38 but in the context of the research presented here, a palmitic acid residue is used as an uncharged substituent with an alkyl side chain.
A key aspect of the research reported herein is solid-phase conjugation of amino acids to ONs. Addition of multiple amino acids to the synthesised ONs was accomplished by solid phase coupling to 2′-amino-LNA monomers already functionalised with one glycyl moiety. This conjugation approach is compatible with both automated phosphoramidite-based ON synthesis and fluoren-9-ylmethoxycarbonyl (Fmoc) based peptide coupling chemistry, and therefore provides effective structural diversification while allowing straight-forward monitoring of reaction yields.
The modified ONs (Fig. 2) were hybridised to complementary DNA or RNA strands and thermal denaturation studies were carried out to assess the duplex-forming ability of the functionalised ONs.
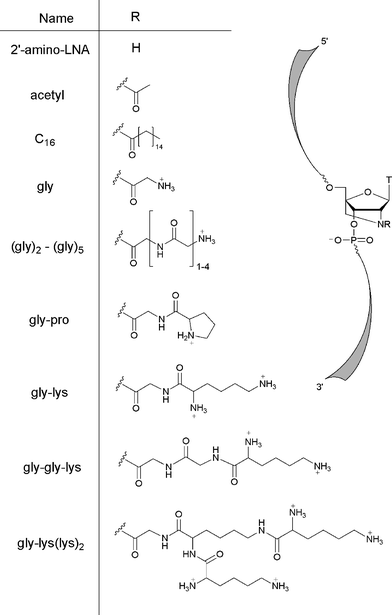 | ||
| Fig. 2 Structures of modified N2′-acyl 2′-amino-LNA monomers. | ||
Results and discussion
Chemistry
Synthesis of the 5′-O-4,4′-dimethoxytritylated 2′-amino-LNA-thymine nucleoside 1 was accomplished according to a literature procedure.39 Selective N2′-acylation of nucleoside 1 was carried out in the presence of O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) as coupling reagent to furnish N-(Fmoc)glycyl derivative 2a. The 2′-N-palmitoyl derivative 2b was obtained by reacting nucleoside 1 and palmitoyl chloride without a coupling reagent (Scheme 1).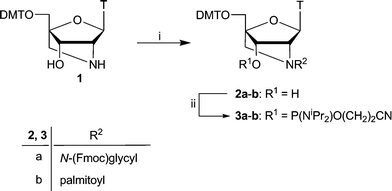 | ||
| Scheme 1 (i) 2a: N-(Fmoc)glycine, DMF, HATU, DIPEA, rt, 1 h, 76%; 2b: palmitoyl chloride, CH2Cl2, pyridine, 0 °C, 2 h, 68%; (ii) 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphane, N,N-diisopropyl-ammonium tetrazolide, CH2Cl2, rt, 12 h (3a: 94%, 3b: 61%). T = thymin-1-yl; DMT = 4,4′-dimethoxytrityl. | ||
The 3′-hydroxy group of nucleoside derivatives 2a–2b was phosphitylated using standard conditions to afford the corresponding phosphoramidite building blocks 3a–3b that were used for automated solid-phase ON synthesis (Scheme 1). Synthesis of the N-acetyl derivative will be published elsewhere,40 whereas literature procedures were used to synthesise the N-trifluoroacetyl protected 2′-amino LNA phosphoramidite.8
Based on DMT cation measurement, ON synthesis proceeded in >90% stepwise coupling yields for modified amidites 3a–b, and in >99% yields for unmodified DNA amidites. In the case of the ONs with the modifications “(gly)2”, “(gly)3”, “(gly)4”, “(gly)5”, “gly-pro”, “gly-lys”, “gly-gly-lys”, and “gly-lys(lys)2”, further conjugation was carried out by solid-phase synthesis after completion of sequences containing a 2′-N-glycyl-2′-amino-LNA monomer (“gly”, see Fig. 2) (vide infra). In the case of the remaining ONs, where no further modifications were required,‡ the ONs were released from the support and purified. The purity of the ONs were determined by ion exchange HPLC (>80%), and their identity confirmed by MALDI-TOF mass spectrometry (for ON sequences, see captions of Tables 1 and 2). For further synthetic details, see experimental section and ESI.†
| DNA modification (charge difference) | Medium salt (110 mM Na+) | |||
|---|---|---|---|---|
| 3′-d(CAC TAT ACG)-5′ | 3′-r(CAC UAU ACG)-5′ | |||
| T m (°C) | ΔTm/modification (°C) | T m (°C) | ΔTm/modification (°C) | |
| Thermal denaturation temperatures were measured using 1.0 μM of each strand of sequence 5′-GTG AXA TGC-3′ and complementary DNA or RNA as shown, where X is the modified monomer. Likewise, 1.0 μM of each strand of sequence 5′-GXG AXA XGC-3′ and complementary DNA or RNA, in the case of those entries prefaced by “3 ×” (e.g. “3 × acetyl”). All Tm values were measured in Na2HPO4/NaH2PO4 buffer with 110 mM Na+ concentration at pH 7. The noted charge difference is relative to the DNA reference and anticipates protonated terminal amino groups of side chains. | ||||
| none (DNA ref.) | 30 | Reference | 27 | Reference |
| 2′-amino-LNA (0) | 36 | +6 | 36 | +9 |
| acetyl (0) | 36 | +6 | 38 | +11 |
| C16 (0) | 31 | +1 | 33 | +6 |
| gly (+1) | 38 | +8 | 37 | +10 |
| (gly)2 (+1) | 38 | +8 | 37 | +10 |
| (gly)3 (+1) | 38 | +8 | 37 | +10 |
| (gly)4 (+1) | 37 | +7 | 38 | +11 |
| (gly)5 (+1) | 36 | +6 | 35 | +8 |
| gly-pro (+1) | 37 | +7 | 38 | +11 |
| gly-lys (+2) | 39 | +9 | 37 | +10 |
| gly-gly-lys (+2) | 41 | +11 | 38 | +11 |
| gly-lys(lys)2 (+4) | 41 | +11 | 39 | +12 |
| 3 × 2′-amino-LNA (0) | 41 | +4 | 49 | +7 |
| 3 × acetyl (0) | 45 | +5 | 53 | +9 |
| 3 × C16 (0) | — | — | 21 | −2 |
| 3 × gly (+3) | 47 | +6 | 51 | +8 |
| DNA modification (charge difference) | Low salt (10 mM Na+) | |||
|---|---|---|---|---|
| 3′-d(CAC TAT ACG)-5′ | 3′-r(CAC UAU ACG)-5′ | |||
| T m/°C | ΔTm/modification (°C) | T m/°C | ΔTm/modification (°C) | |
| Thermal denaturation temperatures were measured using 1.0 μM of each strand of sequence 5′-GTG AXA TGC-3′ and complementary DNA or RNA, where X is the modified monomer. Likewise, 1.0 μM of each strand of sequence 5′-GXG AXA XGC-3′ and complementary DNA or RNA, in the case of those entries prefaced by “3 ×” (e.g. “3 × acetyl”). All Tm values were measured in Na2HPO4/NaH2PO4 buffer with 10 mM Na+ concentration at pH 7. The noted charge difference is relative to the DNA reference, and anticipates protonated terminal amino groups of side chains. | ||||
| none (DNA ref.) | 14 | Reference | 11 | Reference |
| 2′-amino-LNA (0) | 20 | +6 | 19 | +8 |
| acetyl (0) | 20 | +6 | 21 | +10 |
| C16 (0) | 15 | +1 | 18 | +7 |
| gly (+1) | 24 | +10 | 22 | +11 |
| (gly)2 (+1) | 23 | +9 | 22 | +11 |
| (gly)3 (+1) | 23 | +9 | 22 | +11 |
| (gly)4 (+1) | 23 | +9 | 22 | +11 |
| (gly)5 (+1) | 23 | +9 | 22 | +11 |
| gly-pro (+1) | 23 | +9 | 23 | +12 |
| gly-lys (+2) | 27 | +13 | 22 | +11 |
| gly-gly-lys (+2) | 29 | +15 | 25 | +14 |
| gly-lys(lys)2 (+4) | 31 | +17 | 25 | +14 |
| 3 × 2′-amino-LNA (0) | 26 | +4 | 32 | +7 |
| 3 × acetyl (0) | 28 | +5 | 37 | +9 |
| 3 × C16 (0) | — | — | 12 | +0.3 |
| 3 × gly (+3) | 36 | +7 | 37 | +9 |
Solid-phase conjugation
For further acylation of an ON containing a 2′-N-glycyl 2′-amino-LNA monomer incorporated in the middle of a DNA nonamer, the Fmoc-protected glycyl-modified ON was kept on the resin, the amino group of the glycyl moiety deprotected, and then reacted further using standard Fmoc chemistry (Scheme 2, example).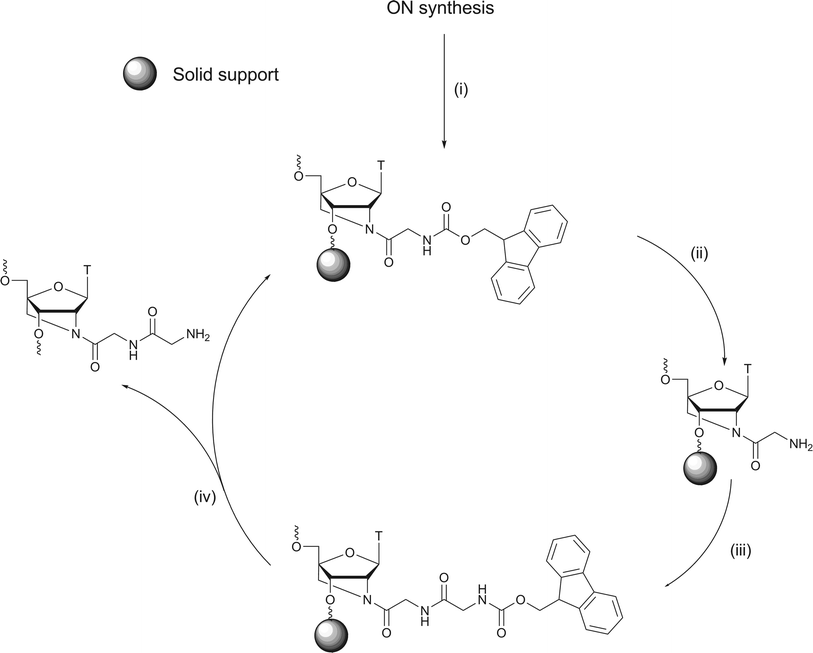 | ||
| Scheme 2 (i) 20% v/v DEA in MeCN, 10 mL, 5 min, rt, (ii) 20% v/v piperidine in DMF, 1.0 mL, rt, 20 min, (iii) N-(Fmoc)-amino acid, DMF, HATU, DIPEA, rt, 3 h, (iv) 28–30% NH3 in water, 1 mL, 55 °C, 12 h. | ||
The 2-cyanoethyl phosphate triester is not stable to Fmoc-deprotection conditions29,41 and was deprotected selectively using diethyl amine (DEA) prior to Fmoc-removal. This eliminates the risk of conjugate addition of the unprotected amino acid amine groups to the acrylonitrile released from phosphate deprotection. Subsequent peptide couplings furnished ONs conjugated with di-, tri-, tetra- or pentapeptides at the N2′-atom of the internally positioned 2′-amino-LNA monomer.
It is possible to monitor the efficiency of the coupling process by measuring the absorbance, and thus the concentration, of the N-(9-fluorenylmethyl)piperidine released upon deprotection of Fmoc-protected amino acids using piperidine.42,43 Most individual peptide couplings proceed in good yields and the overall synthesis yields were estimated to be 5–83%. There is trend that the more solid-phase reactions have to be carried out, the lower the overall yield, and further that the oligonucleotides with charged modifications are isolated in lower yield that those with uncharged modifications (a detailed breakdown of individual yields for different ONs can be found in the ESI). The theoretical loading of the resin used for ON synthesis was used as a starting point and the optical density of the purified ON as an endpoint (see experimental section and ESI for further details).†
Thermal stability studies
We determined the thermal denaturation temperatures (Tm values) of modified nonamer ONs with both DNA and RNA complementary strands in sodium phosphate buffers with concentrations of Na+ of 10 mM (low salt buffer) and 110 mM (medium salt buffer). The results of the thermal stability studies are summarised in Tables 1–4.| Modification X | DNA reference | 2 × LNA | 2 × 2′-amino-LNA | 2 × gly |
|---|---|---|---|---|
| T m/°C | T m/°C | T m/°C | T m/°C | |
| T m values measured in Na2HPO4/NaH2PO4 buffer with 110 mM Na+ concentration at pH 7 using 1.0 μM of each single strand. Numbers in brackets denote change in Tm value per modification relative to the DNA:DNA reference duplex | ||||
| DNA reference | 30 | 41 (+6) | 38 (+4) | 45 (+8) |
| 3 × LNA | 45 (+5) | 62 (+6) | ||
| 1 × 2′-amino-LNA | 36 (+6) | 45 (+5) | ||
| 3 × 2′-amino-LNA | 41 (+4) | 55 (+5) | ||
| 1 × gly | 38 (+8) | 50 (+7) | ||
| 3 × gly | 47 (+6) | 58 (+6) | ||
| Modification X | DNA reference | 2 × LNA | 2 × 2′-amino-LNA | 2 × gly |
|---|---|---|---|---|
| T m/°C | T m/°C | T m/°C | T m/°C | |
| T m values measured in Na2HPO4/NaH2PO4 buffer with 10 mM Na+ concentration at pH 7 using 1.0 μM of each single strand. Numbers in brackets denote change in Tm value per modification relative to the DNA:DNA reference duplex | ||||
| DNA reference | 14 | 25 (+6) | 22 (+4) | 34 (+10) |
| 3 × LNA | 29 (+5) | 45 (+6) | ||
| 1 × 2′-amino-LNA | 20 (+6) | 30 (+5) | ||
| 3 × 2′-amino-LNA | 26 (+4) | 38 (+5) | ||
| 1 × gly | 24 (+10) | 37 (+8) | ||
| 3 × gly | 36 (+7) | 44 (+6) | ||
The ON modified with a glycyl monomer at the 2′-nitrogen (“gly”) (ΔTm = +8 °C) is slightly more stable than unconjugated 2′-amino-LNA (ΔTm = +6 °C). Attaching further glycyl groups by solid-phase conjugation to make di- or tripeptides (“(gly)2” and “(gly)3”) does not alter the melting temperature. For the tetra- and pentapeptides there is a slight decrease in Tm, but further studies are required to determine whether this trend will continue for longer peptide chains.
Steric factors do not appear to greatly influence the stabilisation, since conjugation of the cyclic amino acid proline as the second amino acid (“gly-pro”) has the same effect as glycine (“(gly)2”). Stabilisation does appear to be charge-dependent, as conjugation of a single lysine residue (“gly-lys”) raises the ΔTm to +9 °C, and adding a lysine branch (“gly-lys(lys)2”, four protonated amino groups) gives a ΔTm value of +11 °C.
For comparison, a 2′-amino-LNA monomer was conjugated with palmitic acid at the 2′-nitrogen (“C16”). The C16 and acetyl modifications are comparable in length to the modifications “(gly)5” and “gly”, respectively. The palmitoyl residue especially is obviously significantly more lipophilic and does not offer stabilisation through electrostatic interactions or extensive hydration effects.
For the palmitic acid modification (“C16”), the Tm value was 1 °C higher with a DNA complementary strand versus the unmodified DNA/DNA duplex. Conjugation of 2′-amino-LNA monomers with a palmitic acid residue thus lead to significantly less stabilisation than obtained by unconjugated 2′-amino-LNA or amino acid modified 2′-amino-LNA monomers.
Upon incorporation of three 2′-amino-LNA monomers in the sequence 5′-GXG AXA XGC-3′ (entries “3 × 2′-amino-LNA”, “3 × acetyl” and “3 × gly” in Tables 1 and 2), the affinity effects are nearly additive as will be discussed later. When three palmitoyl-conjugated monomers are incorporated into the nonamer (“3 × C16”), no duplex formation can be seen.
When measuring the thermal stability of duplexes with an RNA complementary strand, very similar results are seen. The relative stabilisation incurred by modification with positively charged residues is a bit larger, “gly”-”(gly)3” has ΔTm values of +10 °C, and “(gly)4” even shows a ΔTm value of +11 °C. Here, the slightly lower stabilisation seen for the longer peptide chains in DNA:DNA duplexes is not apparent except for the pentaglycylated duplex (“(gly)5”) which has a ΔTm value of +8 °C. As for the DNA:DNA duplexes, no steric influence is seen with conjugation of proline, but unlike those, the lysine-modifed ONs show no particular increase in stability compared to the singly charged amino acids. For the modification with four potentially protonated amino groups (“gly-lys(lys)2”), the ΔTm value is +12 °C which is only 1 °C higher than the highest melting temperature for a singly charged modification, and “gly-lys”, with two positive charges, is no different from “gly”. The acetyl modification is under these conditions as stable as the charged modifications. The palmitic acid modification shows a ΔTm value of +6 °C compared to the unmodified DNA reference duplex, again a destabilisation compared to unconjugated 2′-amino-LNA and the charged modifications. For ONs with three modified monomers incorporated, the duplex formed between RNA and the strand with three acetyl-modified 2′-amino-LNA monomers (“3 × acetyl”) is more stable, by 2 °C, than the glycyl duplex (“3 × gly”) making this the most stable of all duplexes, indicating that favourable hydration or steric orientation of the acyl group may be more important than electrostatic effects under these conditions. The ON with three palmitoyl-modifications does form a duplex with RNA, but it is severely destabilised.
The relatively more pronounced stabilising effect with an RNA counter strand is not unexpected, since a similar trend has been observed for LNA modified ONs.5 This RNA-stabilising effect does not appear to be amplified by the electrostatic stabilisation from multiple positive charges.
Measurements have also been carried out in a low salt buffer (10 mM Na+). These measurements do show very similar, but much more pronounced trends compared to the measurements in the medium salt buffer. For duplexes with a DNA counter strand, unconjugated (“2′-amino-LNA”) and acetylated (“acetyl”) 2′-amino-LNA-T modifications stabilise the duplex by +6 °C. In comparison, the singly charged modifications show ΔTm values of +9–10 °C, with no decrease in stabilisation even with longer chains (“(gly)4” and “(gly)5”) or with a cyclic amino acid (“gly-pro”). The charge dependence is evident with the modification “gly-lys” giving a ΔTm value of +13 °C and the branched lysine chain “gly-lys(lys)2” giving a ΔTm value of +17 °C. This is a melting temperature 12 °C higher than seen for the duplex with unmodified 2′-amino-LNA. Although the effect of the multiply charged lysines is once again stronger with a DNA than with an RNA counter strand, the increased stabilisation and charge dependence seen in the low salt buffer supports the hypothesis that stabilisation is facilitated by electrostatic interactions between the positively charged residues and the negatively charged ON backbone. Such an effect is expected to be stronger in the low salt buffer where the concentration of positive counter ions is lowered.
Again, multiple modifications are near additive. A slight decrease in the ΔTm per modification is seen when more modified nucleotides are incorporated. This effect is strongest for duplexes with a DNA complement, especially for the modification “gly”. When three palmitoyl residues are incorporated (“3 × C16”), no melting transition is seen with a DNA complementary strand. While a duplex does form with an RNA complementary strand, no significant stabilisation is seen compared to an unmodified DNA ON. In comparison to unconjugated 2′-amino-LNA, the palmitoyl residues have a destabilising effect.
It is clear from the measurements described above that N2′-acylation of 2′-amino-LNA monomers is an excellent choice for appending amino acids and oligopeptides to ONs. When comparing the effects of the palmitoyl unit with those of the pentaglycyl units, it is clear that hydrophobicity of the conjugated moiety can be unfavorable for duplex stability, an effect which may originate from differences in hydration of the appended side chains.
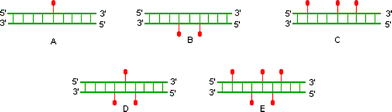 | ||
| Fig. 3 Duplex types measured for thermal stability. Sequences of single strands are 5′-GXG AXA XGC-3′ and the complementary 5′-GCAXAX CAC-3′ where X = DNA-T, LNA-T, 2′-amino-LNA-T or 2′-N-glycylamino-LNA-T (these monomers are shown as droplets). | ||
This study is relevant for several applications. Thus, while antisense applications only require one modified strand in a duplex, aptamers and siRNA constructs may benefit from modifications on both strands, e.g. to stabilise basepairing regions such as stems.
Melting studies were carried out first in a medium salt buffer (Table 3). For all modifications, the stabilisation per modification is close to additive, with only a slight decrease when multiple modifications are present in each strand/duplex. In general, glycyl-modified monomers generate the highest relative thermal stability. The differences in stabilisation between the acylated and unconjugated 2′-amino-LNA are not great, but they are consistent across the various duplex types. Furthermore, the increase in stabilisation is sufficiently large for the charged modification (“gly”) to surpass LNA in stabilisation for duplex types B and C.
The thermal stability of duplexes with modifications in both strands (Fig. 3) has also been studied in a low salt buffer as summarised in Table 4. Here, ΔTm values per modification are increased 1–2 °C for duplexes incorporating the charged glycyl amino group compared to the medium salt buffer. In comparison, the uncharged modifications (“LNA” and “2′-amino-LNA”) show the exact same ΔTm per modification as in the medium salt buffer.
| DNA modification | T m/°C | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3′-d(CAC TBT ACG)-5′ | 3′-r(CAC UBU ACG)-5′ | ||||||||
| B = | A | C | G | T | A | C | G | U | |
| T m values for matched and mismatched duplexes were measured in Na2HPO4/NaH2PO4 buffer at pH 7 (110 mM Na+) using 1.0 μmol of each ON strand. Due to different instrumentation used, there are minor differences in the Tm values reported for some matched duplexes above and in Tables 1 and 3. The ON “3 × C16” is not reported due to the previously mentioned inability to form duplexes with complementary DNA. | |||||||||
| none (DNA ref.) | 28 | 12 | 19 | 13 | 27 | <10 | 22 | 15 | |
| LNA | 34 | 15 | 24 | 17 | 35 | 15 | 28 | 17 | |
| gly | 37 | 20 | 22 | 17 | 38 | 22 | 25 | 19 | |
| C16 | 30 | 16 | 17 | 16 | 33 | 15 | 21 | 15 | |
| 3 × LNA | 43 | 23 | 31 | 26 | 52 | 35 | 42 | 34 | |
| 3 × gly | 46 | 31 | 28 | 26 | 51 | 34 | 38 | 33 | |
Conclusions
In conclusion, we have synthesised phosphoramidite derivatives of a 2′-amino-LNA N2′-acylated with glycine. N-Glycylated 2′-amino-LNA oligonucleotides were further modified by solid phase conjugation to yield di-, tri-, tetra- and pentapeptide modifications as well as an oligonucleotide with a lysine-branched chain. When incorporated into a DNA strand, these amino acid and peptide modifications increase stability of duplexes formed with both DNA and RNA complements. The stabilising effect in general becomes more profound when thermal denaturation studies are carried out in a buffer with low counterion concentration. A duplex containing 2′-amino-LNA monomer(s) modified with positively charged amino acids is more stable than DNA/DNA or DNA/RNA duplexes and more stable than the corresponding duplexes with unconjugated 2′-amino-LNA monomer(s) incorporated. The stabilising effect was found to be nearly additive for multiple incorporations of modified monomers in one or both strands.Furthermore, oligonucleotides containing 2′-amino-LNA monomers N-acylated with palmitic or acetic acid were synthesised. Unlike the amino acid modifications, only negligible changes in thermal stability were observed for these modifications when the counterion concentration of the buffer was decreased. We propose that the stabilising influence of the amino acid-modified monomers is due to an electrostatic effect. The positively charged protonated amino groups of one strand may interact with the negatively charged phosphate backbone of the same strand, decreasing the effective charge of that strand and thus the repulsive electrostatic interaction with the negatively charged complementary strand. It is also possible that positively charged amino groups of one strand interact with negatively charged phosphate groups on the complementary strand, creating an attractive electrostatic interaction.
N2′-Acylated 2′-amino-LNA monomers are appealing as constituents of biologically active ONs, e.g.antisense, aptamer or siRNA constructs, as they offer an opportunity of increasing molecular diversity and modulating physicochemical properties while preserving high-affinity DNA and RNA-binding.
Experimental section
General
All reagents were used as purchased without further purification. Reactions (except solid-phase conjugation of amino acids) were conducted under an atmosphere of nitrogen. Petroleum ether, used for column chromatography, was of distillation range 60–80 °C. For column chromatography, silica gel 60 (0.040–0.063 mm) from Merck was used. Progress in reactions was monitored by thin-layer chromatography (TLC) on analytical silica gel TLC plates (60 F254) from Merck with UV-light used for visualisation. NMR spectra were recorded on a Varian Gemini 2000 NMR spectrometer. The δ values are in ppm relative to tetramethylsilane as internal standard for 1H NMR, deuterated solvent CDCl3 (δ 77.00) for 13C NMR, and relative to 85% H3PO4 as external standard for 31P NMR. Assignments of NMR spectra, when given, are based on 2D spectra and follow the standard carbohydrate/nucleoside nomenclature (the carbon atom of the C4′-substituent is numbered C5′′). The assignments of methylene protons, when given, may be interchanged. Coupling constants (J-values) are given in Hertz. The NMR spectra of the N-acylated 2′-amino-LNA nucleosides revealed two conformations of the amide bond (rotamers). High resolution MALDI spectra were recorded in positive ion mode on a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer (IonSpec, Irvine, CA). For accurate ion mass determinations, the [M + Na]+ ion was peak matched using ions derived from the 2,5-dihydroxybenzoic acid matrix. Electrospray mass spectrometry was performed using a Q-Star Pulsar Mass Spectrometer (Sciex/Applied Biosystems) equipped with a nano electrospray source. The instrument was externally calibrated in the relevant range by observing cluster ions (NaI)nNa+. Samples were dissolved to a concentration of approximately 10 μM in methanol and sprayed from metal coated borosilicate emitters.(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-5-[N-(fluoren-9-ylmethoxycarbonyl)glycyl]-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (2a)
Fmoc-gly-OH (312 mg, 1.05 mmol, 1.2 eq), DMF (10 mL) and HATU (400 mg, 1.05 mmol, 1.2 eq) were mixed, DIPEA (0.275 mL, 1.58 mmol, 1.8 eq) was added and the reaction mixture was stirred for 10 min at room temperature. A solution of nucleoside 1 (500 mg, 0.88 mmol) in DMF (10 mL) was added dropwise, and the reaction was stirred for a further 60 min at which point it was deemed complete by TLC. The reaction mixture was diluted with ethyl acetate (300 mL), washed with H2O (2 × 450 mL), 5% NaHCO3 (2 × 450 mL), and finally H2O (2 × 450 mL). The aqueous phases were back-extracted in portions with ethyl acetate (300 mL in total). The combined organic phase was dried (MgSO4), filtered, and the solvent removed in vacuo to yield the crude product as a pale yellow foam (667 mg). The crude product was purified by column chromatography on silica with 60–100% ethyl acetate in petroleum ether, the relevant fractions combined and the eluent removed in vacuo to yield a rotameric mixture (∼0.66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.33 by 1H NMR) of nucleoside 2a as a pale yellow foam (567 mg, 76%). δH (300 MHz, CDCl3, Me4Si) (subscript A = major rotamer, subscript B = minor rotamer; integrals are shown only for dominating rotamer) 9.83 (1H, br s, NHA), 9.43 (br s, NHB), 7.70–7.12 (17H, m, Ar), 7.59 (1H, s, 6-HA), 7.57 (s, 6-HB), 6.80–6.76 (4H, m, Ar), 5.95 (1H, t, 3′-OH), 5.50 (s, H-1′B), 5.44 (1H, s, H-1′A), 5.16 (s, H-2′B), 4.73 (1H, s, H-2′A), 4.38 (1H, s, H-3′), 4.26–4.08 (5H, m, H-5′, H-5′, CH-Fmoc, CH2-Fmoc), 3.71 (6H, s, 2 × OCH3), 3.61–3.45 (4H, m, CH2, H5′, H-5′), 1.97 (s, 5-MeB), 1.58 (3H, s, 5-MeA); δC (75 MHz, CDCl3) 168.0, 164.5, 158.6, 157.0, 150.7, 144.5, 144.4, 143.9, 143.7, 141.2, 135.5, 135.4, 135.3, 135.2, 134.6, 130.1, 130.0, 129.2, 128.1, 127.9, 127.8, 127.7, 127.1, 125.2, 119.9, 113.4, 113.2, 110.7, 88.7, 88.0, 87.1, 86.8, 86.7, 70.1, 69.1, 67.4, 67.3, 63.0, 59.2, 55.2, 51.6, 47.1, 46.9, 43.1, 42.9, 12.7, 12.6; HR ESI MSm/z 873.3070 ([M + Na]+, C49H46N4O10Na+ calcd 873.3106).
:0.33 by 1H NMR) of nucleoside 2a as a pale yellow foam (567 mg, 76%). δH (300 MHz, CDCl3, Me4Si) (subscript A = major rotamer, subscript B = minor rotamer; integrals are shown only for dominating rotamer) 9.83 (1H, br s, NHA), 9.43 (br s, NHB), 7.70–7.12 (17H, m, Ar), 7.59 (1H, s, 6-HA), 7.57 (s, 6-HB), 6.80–6.76 (4H, m, Ar), 5.95 (1H, t, 3′-OH), 5.50 (s, H-1′B), 5.44 (1H, s, H-1′A), 5.16 (s, H-2′B), 4.73 (1H, s, H-2′A), 4.38 (1H, s, H-3′), 4.26–4.08 (5H, m, H-5′, H-5′, CH-Fmoc, CH2-Fmoc), 3.71 (6H, s, 2 × OCH3), 3.61–3.45 (4H, m, CH2, H5′, H-5′), 1.97 (s, 5-MeB), 1.58 (3H, s, 5-MeA); δC (75 MHz, CDCl3) 168.0, 164.5, 158.6, 157.0, 150.7, 144.5, 144.4, 143.9, 143.7, 141.2, 135.5, 135.4, 135.3, 135.2, 134.6, 130.1, 130.0, 129.2, 128.1, 127.9, 127.8, 127.7, 127.1, 125.2, 119.9, 113.4, 113.2, 110.7, 88.7, 88.0, 87.1, 86.8, 86.7, 70.1, 69.1, 67.4, 67.3, 63.0, 59.2, 55.2, 51.6, 47.1, 46.9, 43.1, 42.9, 12.7, 12.6; HR ESI MSm/z 873.3070 ([M + Na]+, C49H46N4O10Na+ calcd 873.3106).
(1R,3R,4R,7S)-1-(4,4′-Dimethoxytrityloxymethyl)-5-(hexadecanoyl)-7-hydroxy-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (2b)
Nucleoside 1 (1.51 g, 2.64 mmol) was co-evaporated with anhydrous toluene (2 × 2 mL) and subsequently dissolved in anhydrous CH2Cl2 (25 mL). Anhydrous pyridine (1.0 mL, 12.4 mmol, 4.7 equiv.) was added, the solution was cooled to 0 °C, and palmitoyl chloride (0.75 g, 2.73 mmol, 1.02 equiv.) was added dropwise. The reaction mixture was stirred at 0 °C for 2 h, whereupon it was diluted with CH2Cl2 (25 mL) and washed with sat. aq. NaHCO3 (2 × 25 mL). The organic phase was dried (MgSO4) and concentrated to dryness under reduced pressure. The residue was purified by silica gel column chromatography using MeOH/pyridine/CH2Cl2 (4:1:95, v/v/v) as eluent to afford a rotameric mixture (∼0.45:0.55 by 1H NMR) of nucleoside 2b as a white solid (1.45 g, 68%). Rf 0.52 (MeOH–CH2Cl2 8:92); δH (300 MHz, CDCl3, Me4Si) (subscript A = major rotamer, subscript B = minor rotamer; integrals are shown only for major rotamer) 9.86 (br s, NHB), 9.44 (1H, br s, NHA), 7.66 (1H, s, 6-HA), 7.60 (s, 6-HB), 7.49–7.20 (9H, m, Ar), 6.84 (4H, m, Ar), 5.53 (s, H-1′B), 5.44 (1H, s, H-1′A), 5.20 (s, H-3′B), 4.53 (1H, s, H-3′A), 4.37 (1H, s, H-2′A), 4.30 (s, H-2′B), 3.78 (6H, s, 2 × OCH3), 3.82–3.62 (4H, m, H-5′, H-5′), 2.50–2.37, 2.17–2.13 (2H, m, CH2), 1.61 (3H, 2 × s, 5-MeA+B), 1.65–1.50 (2H, m, CH2), 1.24–1.22 (24H, m, CH2), 0.87 (3H, t, J = 6.4 Hz, Me); δC (75 MHz, CDCl3) 173.5, 173.2, 164.5, 164.2, 158.7, 158.6, 150.1, 149.7, 144.4, 144.3, 135.5, 135.3, 135.2, 134.7, 130.1, 130.1, 130.0, 128.1, 128.0, 127.1, 127.0, 113.3, 113.2, 110.5, 109.9, 88.7, 88.0, 86.9, 86.8, 86.7, 70.2, 68.9, 63.6, 59.1, 59.6, 55.2, 34.2, 33.9, 31.9, 29.7, 29.6, 29.5, 29.3, 25.0, 24.6, 22.7, 14.1, 12.5, 12.4; HR MALDI MSm/z 832.4475 ([M + Na]+, C48H63N3O8Na+ calcd 832.4507).
(1R,3R,4R,7S)-7-[2-Cyanoethoxy(diisopropylamino)phosphinoxy]-1-(4,4′-dimethoxytrityloxymethyl)-5-[N-(fluoren-9-ylmethoxycarbonyl)glycyl]-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (3a)
Nucleoside 2a (517 mg, 0.61 mmol) was co-evaporated with anhydrous dichloroethane. Diisopropylammonium tetrazolide (209 mg, 1.22 mmol, 2 eq.) was added and the solids dissolved in CH2Cl2 (7 mL). 2-Cyanoethyl-N,N,N′,N′-tetraisopropylphosphane (388 μL, 1.22 mmol, 2.0 eq) was added dropwise and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was diluted with ethyl acetate (2.5 mL), washed with NaHCO3 (20 mL), and the aqueous phase was back-extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (Na2SO4), filtered, and the eluent removed in vacuo, yielding a pale yellow foam (crude yield 866 mg). The foam was dissolved in the minimal amount of ethyl acetate (2 mL) and added dropwise to cold petroleum ether (200 mL, app. 0 °C). The precipitating mixture was stirred for 10 min, the resulting cream-coloured solid filtered off and dried under vacuum overnight. The mother liquor was concentrated in vacuo, the crystallization procedure repeated, and following TLC analysis, the crystallization products were combined (601 mg, 94%). δP (121.5 MHz, CDCl3) 150.0, 149.7, 149.4, 148.3; HR ESI MSm/z 1051.4390 ([M + H]+, C58H64N6O11P+ calcd 1051.4365).(1R,3R,4R,7S)-7-(2-Cyanoethoxy(diisopropylamino)phosphinoxy)-1-(4,4′-dimethoxytrityloxymethyl)-5-(hexadecanoyl)-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (3b)
Nucleoside 2b (1.423 g, 1.76 mmol) and diisopropylammonium tetrazolide (0.453 g, 2.65 mmol, 1.5 equiv.) were mixed and dissolved in anhydrous CH2Cl2 (25 mL). 2-Cyanoethyl-N,N,N′,N′-tetraisopropylphosphane (1.060 g, 3.52 mmol, 2.0 equiv.) was added dropwise and the resulting mixture was stirred at room temperature for 12 h. It was diluted with CH2Cl2 (25 mL), washed with sat. aq. NaHCO3 (2 × 25 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using toluene–EtOH/Et3N (91:7:2, v/v/v) as eluent to afford the amidite 3b as off-white foam (1.089 g, 61%). Rf 0.43 (EtOH–toluene 1:10); δP (121.5 MHz, CDCl3) 149.8, 149.3, 148.8, 147.3; HR ESI MSm/z 1032.5501 ([M + Na]+, C57H80N5O9PNa+ calcd 1032.5586).
Oligonucleotide synthesis and purification
The oligonucleotides were synthesised in the DMT-ON mode on an automated DNA synthesizer using an Expedite™ Nucleic Acid Synthesis System model 8909 from Applied Biosystems. Synthesis was carried out in 0.2–1.0 μmol scale using polystyrene resin purchased from Glen Research. Phosphoramidites 3a–b were dissolved in acetonitrile as a 0.05 M solution and coupled using tetrazole as an activator. The coupling time was 15–20 min for amidites 3a–b, while commercial DNA phosphoramidites were subjected to 2 min coupling time. After completion of the coupling steps, the cyanoethyl protecting groups were removed from the phosphate backbone (20% v/v diethylamine in acetonitrile, 10 mL, 5 min, rt). The resin was washed with acetonitrile (3 × 5 mL) and dried briefly. Oligomers needing no further modification were deprotected and cleaved from the resin using standard conditions (28–30% aqueous ammonia, 1 mL, 12 h, 55 °C) and purified by reversed phase HPLC on a Waters 600 system (with Waters 2996 PDA detector) equipped with a Waters Xterra™ MS C18 10 μm 7.8 × 150 mm column using a 38 min linear gradient of 0–53% MeCN in 0.05 M triethyl ammonium acetate (pH 7.4) at a flow rate of 2.50 mL min−1. The purified oligonucleotides were subjected to standard detritylation and then precipitated from ethanol (“C16”, “3 × C16”) or acetone (all others). The ONs were >80% pure by ion-exchange HPLC (Merck Hitachi D-7000 LaChrom Interface (with L-7400 UV detector) equipped with a Gen-Pak Fax 4.6 × 100 mm column). The composition of the oligonucleotides was verified by MALDI-TOF MS, which was recorded on a Bruker Daltonics Microflex LT using a 3-hydroxy picolinic acid matrix.†General procedure for solid-phase modification of ONs
The 2′-amino group of the 2′-N-glycyl-2′-amino-LNA monomer in the oligomer was deprotected (20% v/v piperidine in DMF, 1.0 mL, 20 min, rt). The liquid phase was removed from the resin and its absorbance measured at 301 nm (100 μL in 900 μL DMF, zeroing the instrument on DMF). The resin was washed successively with DMF (2 × 1.0 mL) and ethanol (2 × 1.0 mL). Fmoc-protected amino acid (30 equivalents), HATU (30 equivalents) and DIPEA (60 equivalents) were mixed in anhydrous DMF (500 μL) and added to the resin. The resulting suspension was shaken gently for 3 h at rt. The liquid phase was removed and the solid support washed with anhydrous DMF (4 × 0.5 mL) and ethanol (2 × 1.0 mL). If required, the procedure was repeated for stepwise addition of further amino acids. When synthesis was complete, the oligomer was cleaved from the resin and deprotected as described above. Purification was carried out either as described above or by simple detritylation and precipitation from acetone or ethanol without HPLC purification when the purity of the crude oligomer was sufficiently high (>80% by ion-exchange HPLC analysis). By measurement of the absorbance of the Fmoc deprotection product (ε301 nm = 7800 M−1 cm−1),43 the solid phase acylation reactions were determined to proceed in good individual yields (data not shown). The overall yield of all modified oligonucleotides was calculated from their UV absorbance at 260 nm (OD260). The final yields were 5–32% for post-synthetically conjugated oligos, and 8–83% for ONs with no further conjugation after automated ON synthesis.†Thermal denaturation experiments
Thermal denaturation studies were carried out on a Varian Cary 100 Bio UV-Visible spectrophotometer with a Peltier controlled 6 × 6 sample changer and Cary WinUV software. Melting temperatures (Tm values) were measured as the maximum of the first derivative of the melting curve (A260vs. temperature). Concentrations and buffers as described in the captions for Tables 1 and 2. For the medium salt buffer, temperature was increased from 8 to 80 °C; increase 1 °C min−1. For the low salt buffer, temperature was increased from 6 to 65 °C; increase 1 °C min−1. Tm values were determined in two independent experiments, consistent within 1 °C, and ΔTm values were calculated per modification relative to the Tm values of the reference duplexes. Mismatch discrimination data were obtained on a Perkin Elmer UV-visible spectrometer Lambda 20 equipped with a PTP-6 Peltier Temperature Programmer.Acknowledgements
The Danish National Research Foundation is thanked for funding the Nucleic Acid Center, a research center for studies on nucleic acid chemical biology. This work was furthermore supported by the Sixth Framework Programme Marie Curie Host Fellowship for Early Stage Research Training under contract number MEST-CT-2004-504018.Notes and references
- C. Wilson and A. D. Keefe, Curr. Opin. Chem. Biol., 2006, 10, 607–614 CrossRef CAS.
- E. T. Kool, Acc. Chem. Res., 2002, 35, 936–943 CrossRef CAS.
- J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265–4274 RSC.
- J. H. Chan, S. Lim and W. S. Wong, Clin. Exp. Pharmacol. Physiol., 2006, 33, 533–540 CrossRef CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- S. Obika, D. Nanbu, Y. Hari, J. Andoh, K. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401–5404 CrossRef CAS.
- S. K. Singh, R. Kumar and J. Wengel, J. Org. Chem., 1998, 63, 6078–6079 CrossRef CAS.
- S. K. Singh, R. Kumar and J. Wengel, J. Org. Chem., 1998, 63, 10035–10039 CrossRef CAS.
- H. Aurup, T. Tuschl, F. Benseler, J. Ludwig and F. Eckstein, Nucleic Acids Res., 1994, 22, 20–24 CAS.
- N. Kalra, M. C. Parlato, V. S. Parmar and J. Wengel, Bioorg. Med. Chem. Lett., 2006, 16, 3166–3169 CrossRef CAS.
- J. Kurreck, Angew. Chem., Int. Ed., 2009, 48, 1378–1398 CrossRef CAS.
- N. Kumar, A. Patowary, S. Sivasubbu, M. Petersen and S. Maiti, Biochemistry, 2008, 47, 13179–13188 CrossRef CAS.
- G. Mayer, Angew. Chem., Int. Ed., 2009, 48, 2672–2689 CrossRef CAS.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham Jr., D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS.
- R. Noir, M. Kotera, B. Pons, J. S. Remy and J. P. Behr, J. Am. Chem. Soc., 2008, 130, 13500–13505 CrossRef CAS.
- N. M. Bell and J. Micklefield, ChemBioChem, 2009, 10, 2691–2703 CrossRef CAS.
- S. H. Weisbrod and A. Marx, Chem. Commun., 2008, 5675–5685 RSC.
- H. Lönnberg, Bioconjugate Chem., 2009, 20, 1065–1094 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- A. de Mesmaeker, K. H. Altmann, A. Waldner and S. Wendeborn, Curr. Opin. Struct. Biol., 1995, 5, 343–355 CrossRef CAS.
- T. Bryld and C. Lomholt, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1645–1647 CrossRef CAS.
- M. Manoharan, K. L. Tivel, T. P. Condon, L. K. Andrade, I. Barber-Peoch, G. Inamati, S. Shah, V. Mohan, M. J. Graham, C. F. Bennett, S. T. Crooke and P. D. Cook, Nucleosides, Nucleotides Nucleic Acids, 1997, 16, 1129–1138 CAS.
- F. Said Hassane, A. F. Saleh, R. Abes, M. J. Gait and B. Lebleu, Cell. Mol. Life Sci., 2010, 67, 715–726 CrossRef CAS.
- M. Antopolsky, E. Azhayeva, U. Tengvall, S. Auriola, I. Jääskeläinen, S. Rönkkö, P. Honkakoski, A. Urtti, H. Lönnberg and A. Azhayev, Bioconjugate Chem., 1999, 10, 598–606 CrossRef CAS.
- F. Debart, S. Abes, G. Deglane, H. M. Moulton, P. Clair, M. J. Gait, J.-J. Vasseur and B. Lebleu, Curr. Top. Med. Chem., 2007, 7, 727–737 CrossRef CAS.
- R. L. Letsinger, C. N. Singman, G. Histand and M. Salunkhe, J. Am. Chem. Soc., 1988, 110, 4470–4471 CrossRef CAS.
- M. Manoharan, K. L. Tivel, L. K. Andrade and P. D. Cook, Tetrahedron Lett., 1995, 36, 3647–3650 CrossRef CAS.
- S. Chaturvedi, T. Horn and R. L. Letsinger, Nucleic Acids Res., 1996, 24, 2318–2323 CrossRef CAS.
- R. H. Griffey, B. P. Monia, L. L. Cummins, S. Freier, M. J. Greig, C. J. Guinosso, E. Lesnik, S. M. Manalili, V. Mohan, S. Owens, B. R. Ross, H. Sasmor, E. Wancewicz, K. Weiler, P. D. Wheeler and P. D. Cook, J. Med. Chem., 1996, 39, 5100–5109 CrossRef CAS.
- N. Kalra, B. R. Babu, V. S. Parmar and J. Wengel, Org. Biomol. Chem., 2004, 2, 2885–2887 RSC.
- M. D. Sørensen, M. Petersen and J. Wengel, Chem. Commun., 2003, 2130–2131 RSC.
- T. Umemoto, J. Wengel and A. S. Madsen, Org. Biomol. Chem., 2009, 7, 1793–1797 RSC.
- M. Kalek, A. S. Madsen and J. Wengel, J. Am. Chem. Soc., 2007, 129, 9392–9400 CrossRef CAS.
- T. Umemoto, P. J. Hrdlicka, B. R. Babu and J. Wengel, ChemBioChem, 2007, 8, 2240–2248 CrossRef CAS.
- D. Lindegaard, A. S. Madsen, I. V. Astakhova, A. D. Malakhov, B. R. Babu, V. A. Korshun and J. Wengel, Bioorg. Med. Chem., 2008, 16, 94–99 CrossRef CAS.
- P. J. Hrdlicka, B. R. Babu, M. D. Sørensen and J. Wengel, Chem. Commun., 2004, 1478–1479 RSC.
- P. J. Hrdlicka, B. R. Babu, M. D. Sørensen, N. Harrit and J. Wengel, J. Am. Chem. Soc., 2005, 127, 13293–13299 CrossRef CAS.
- N. N. Polushin and J. S. Cohen, Nucleic Acids Res., 1994, 22, 5492–5496 CrossRef CAS.
- C. Rosenbohm, S. M. Christensen, M. D. Sørensen, D. S. Pedersen, L.-E. Larsen, J. Wengel and T. Koch, Org. Biomol. Chem., 2003, 1, 655–663 RSC.
- B. R. Babu, M. C. Wamberg, J. Maity, T. B. Jensen, M. D. Sørensen, I. V. Astakhova, A. D. Malakhov, T. S. Kumar, D. Lindegaard, N. Kalra, V. A. Korshun, P. J. Hrdlicka, A. K. Prasad, V. S. Parmar and J. Wengel, manuscript in preparation.
- J. Nielsen and K. D. Janda, Methods, 1994, 6, 361–371 CrossRef CAS.
- L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404–3409 CrossRef CAS.
- S. K. Shannon and G. Barany, J. Org. Chem., 2004, 69, 4586–4594 CrossRef CAS.
- O. Plashkevych, S. Chatterjee, D. Honcharenko, W. Pathmasiri and J. Chattopadhyaya, J. Org. Chem., 2007, 72, 4716–4726 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: MALDI TOF mass spectrometry data for modified ONs, overview of yields for synthesis of modified ONs, 1H and 13C NMR spectra for compounds 2a–b, 31P NMR spectra for compounds 3a–b. See DOI: 10.1039/c0ob00532k |
| ‡ “2′-amino-LNA”, “acetyl”, “gly”, “3 × 2′-amino-LNA”, “3 × acetyl”, “3 × gly”, “C16” and “3 × C16” |
| This journal is © The Royal Society of Chemistry 2011 |