The traceless Staudinger ligation for indirect 18F-radiolabelling†‡
Laurence
Carroll
a,
Sophie
Boldon
a,
Romain
Bejot
a,
Jane E.
Moore
b,
Jérôme
Declerck
c and
Véronique
Gouverneur
*a
aUniversity of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk
bAstraZeneca UK, Alderley Park, Cheshire, UK SK10 4TG
cSiemens Molecular Imaging, 23–38 Hythe Bridge Street, Oxford, OX1 2EP, UK
First published on 22nd November 2010
Abstract
The Staudinger ligation of phosphine-substituted thioesters with 18F-fluoroethylazide has been successfully applied to access 18F-labelled molecules in radiochemical yields superior to 95%; the first fluorous variant of a Staudinger radio-ligation has been validated.
Introduction
Two transformations involving the azide group currently dominate bioorthogonal chemistry, the azide-alkyne 1,3-dipolar cycloaddition leading to triazoles, and the Staudinger ligation, a process forming an amide bond by reacting an azide with a phosphine-substituted ester.1 Both reactions have appealing features to study biomolecules in their native settings and have found endless applications for labelling and beyond.2 The ability to radiolabel biomarkers with the nuclide 18F is essential to advance positron emission tomography (PET), an imaging modality used to conduct diagnostic clinical studies, understand biochemical processes and support drug development programmes.3 The short half-life of positron-emitting fluorine-18 (t½ = 109.7 min) demands rapid and efficient methodology to access [18F]-labelled biomarkers in high radiochemical yield and purity.4 Fluorine-labelling of complex biomolecules such as peptides, antibodies or oligonucleotides remains challenging and is not always compatible with existing direct 18F-labelling strategies. Methods have therefore been developed that employ non carrier-added 18F-labelled prosthetic groups for chemoselective coupling under mild reaction conditions.4 For this multi-step indirect labelling approach, it is of the utmost importance for the coupling process to be highly efficient and for the new functionality installed upon labelling, not to perturb the physicochemical properties or in vivo behavior of the 18F-labelled biomarker. Copper-catalysed cycloaddition with either the alkyne or the azide carrying the 18F-tag has been successfully applied for labelling small molecules and peptides.5 The process generates a 1,2,3-triazole motif, a convenient bioisosteric replacement of the amide bond but the toxicity of copper(I) is limiting for some in vivo applications. The so-called traceless Staudinger ligation is a highly attractive process leading to a native amide bond without inclusion of the phosphine oxide in the final product.6 The synthetic value of this remarkable transformation has been demonstrated by Raines and coworkers with the ligation of complex peptide fragments, possibly best illustrated with the total synthesis of ribonuclease A.7Herein, we report the first examples of Staudinger ligation with 2-[18F]fluoroethylazide 1 for the 18F-labelling of small molecules through formation of an amide bond. For this study, we selected the traceless Staudinger ligation of a phosphinothioester with an 18F-labelled azide fragment. Mechanistically, the attack of the phosphine onto the azide leads to an iminophosphorane, which rearranges to afford an amidophosphonium salt. After hydrolysis, the labelled amide is formed with concomitant release of the phosphine oxide (Scheme 1). This transformation benefits from the absence of metal catalyst but its value in the context of radiolabelling relies on the validation of a protocol with favorable reaction kinetics, and which allows for easy separation of the 18F-labelled amide from the phosphine oxide by-product.
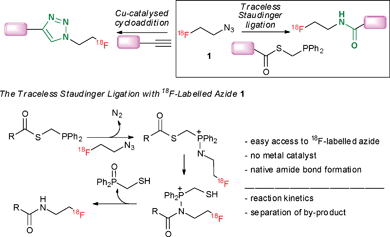 | ||
| Scheme 1 18F-labelled azide 1 as a prosthetic group for indirect labelling. | ||
Results and discussion
The borane-protected phosphinothiol 2 was chosen as a common building block to access masked (diphenylphosphino)methanethiol thioesters, which after deprotection, could undergo traceless Staudinger ligation with the azide 1. Deprotonation of the commercially available (diphenylphosphonio)trihydroborate complex with sodium hydride in DMF at 0 °C followed by alkylation with (bromomethyl)ethanethioate 3 (easily prepared in two steps from thioacetic acid), gave the doubly protected trihydroborate complex 4 in a moderate yield of 60%.8 Treatment of 4 with sodium methoxide in methanol for ten minutes at room temperature revealed the borane-protected thiol moiety 2. This thiol was engaged in the next step without purification (Scheme 2, Stage 1). Coupling of 2 with different carboxylic acids was undertaken using standard DCC conditions. Subsequent removal of the borane protecting group by treating the coupled adduct with 1,4-diazabicyclo [2.2.2]octane (DABCO) in toluene at 60 °C for 1 h afforded adducts 5 in moderate yields over two steps (Scheme 2, Stage 2).9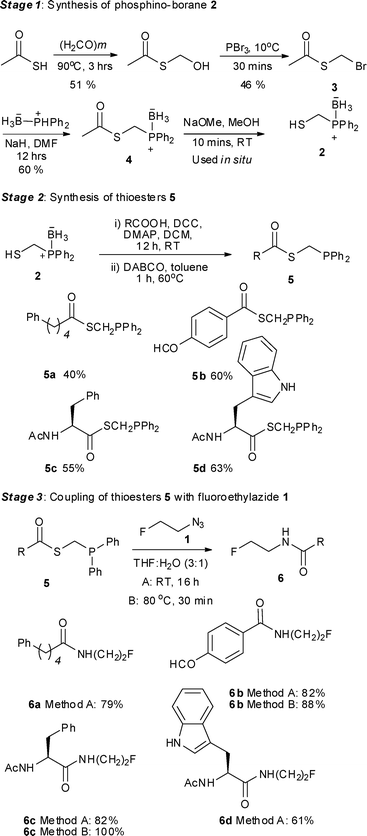 | ||
| Scheme 2 Staudinger ligation with fluorinated azide 1. | ||
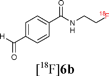
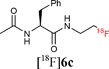
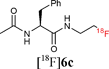
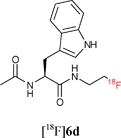
Starting from 5-phenylpentanoic acid, 5a could be accessed in 40% overall yield. Compound 5b was synthesised in 60% yield over two steps and starting from N-acetylphenylalanine or N-acetyltryptophan, 5c and 5d were accessed in 55% and 63% overall yield respectively. The Staudinger ligation of 5a–d with 1 in THF–H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under standard ligation conditions (RT, 16 h) gave the desired fluorinated targets 6 in moderate to excellent yields (Scheme 2, Stage 3, Method A). Reacting 5a and 5b with 1 accessed 6a and 6b in isolated yields of 79% and 82% respectively. Staudinger ligation of the (L)-phenylalanine-derived precursor 5c with 1 gave the fluorinated amide 6c in 82% yield. The ligation of 5d was slightly less efficient as the product was contaminated with the starting material both as free phosphine and phosphine oxide. Since the long reaction times employed in these initial studies are not compatible with fluorine-18 radiolabelling, further optimisation was undertaken (Scheme 2, Stage 3, Method B). Heating the ligation mixture at 80 °C enabled the reaction time to be reduced to 30 min with conversion up to 100%.
:1) under standard ligation conditions (RT, 16 h) gave the desired fluorinated targets 6 in moderate to excellent yields (Scheme 2, Stage 3, Method A). Reacting 5a and 5b with 1 accessed 6a and 6b in isolated yields of 79% and 82% respectively. Staudinger ligation of the (L)-phenylalanine-derived precursor 5c with 1 gave the fluorinated amide 6c in 82% yield. The ligation of 5d was slightly less efficient as the product was contaminated with the starting material both as free phosphine and phosphine oxide. Since the long reaction times employed in these initial studies are not compatible with fluorine-18 radiolabelling, further optimisation was undertaken (Scheme 2, Stage 3, Method B). Heating the ligation mixture at 80 °C enabled the reaction time to be reduced to 30 min with conversion up to 100%.
In addition to thioesters 5a–d, we also prepared the fluorous derivative 7 (Scheme 3).10 The fluorodetagging of fluorous precursors has recently been validated using direct 18F-fluorination,11–13 however detagging upon displacement with a 18F-labelled prosthetic group has not been reported. The Staudinger ligation is an ideal platform for proof of concept as the fluorous tag can be attached onto the phosphino fragment of the starting thioester.14 The fluorous-tagged thioester 7 bearing two C4F9 tags on the phosphine was therefore synthesised following a reaction sequence similar to the one applied for the preparation of non fluorous thioesters. This compound was found to oxidise to the corresponding phosphine oxide faster than the non fluorous counterpart. The extent of oxidation was minimised during purification by performing the silica gel chromatography under an atmosphere of N2. Staudinger ligation with 1 using standard conditions led to the formation of 6c in 27% yield with purification of the reaction mixture using FSPE. The decrease in isolated yield could be attributed to the propensity of the phosphane precursor 7 towards oxidation.
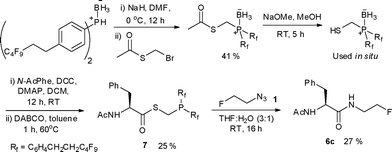 | ||
| Scheme 3 Fluorous Staudinger ligation with 1. | ||
With key studies of the Staudinger ligation with the fluorinated azide 1 in hand, focus turned to the validation of a radiochemical variant of this approach using 2-[18F]fluoroethylazide 1. This 18F-labelled azide was prepared by nucleophilic fluorination of the fluorous-tagged precursor 2-azido-ethyl-1H,1H,2H,2H-perfluorodecane-1-sulfonate. The fluorination was performed in acetonitrile using [18F]-/K+-Kryptofix 222 and fluorous solid phase extraction (FSPE) implemented to separate the radiolabelled product [18F]1 from the excess fluorous precursor (specific activity of ∼10 GB q μmol−1).11 This protocol is superior to previous radiosyntheses that rely on purification by distillation. The FSPE-purified azide [18F]1 was recovered in a mixture of H2O and CH3CN. The ligations were performed adding [18F]1 to a solution of the thioester dissolved in THF–H2O (4:1) followed by heating at 80 °C for 30 min. The reaction time could be reduced to 15 min by increasing the temperature to 120 °C (Method C). Under these conditions, the ligation of 5a–b with [18F]1 proceeded in excellent radiochemical yields superior to 95% (entries 1–2, Table 1). The reactions were equally successful when performed in a mixture of DMF–H2O (6:1) (Method D).15 In contrast to couplings carried out in THF, the efficiency of the ligation process in DMF was not found to be dependent on concentration. Pleasingly, the labelling of both the non fluorous and fluorous alanine derivative 5c and 7 with [18F]1 in DMF–H2O (6:1) at 120 °C for 15 min afforded [18F]6c in an excellent RCY in excess of 95% (entries 3–4, Table 1). For the ligation of 7, purification using FSPE led to an improvement in chemical purity although breakthrough of fluorous material into the fluorophobic eluted fraction was seen. HPLC was therefore necessary to obtain analytically pure sample of the 18F-labelled amide. The ligation of the tryptophan derivative 5d was also very high yielding with the totality of the azide converted into the 18F-labelled amide (entry 5, Table 1). The overall synthesis duration was within 30 min. Pleasingly, all radio-HPLC traces typically showed only the presence of the ligated product in crude reaction mixture (Fig. 1).
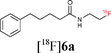
![Radio-HPLC trace of crude material [18F]6a.](/image/article/2011/OB/c0ob00564a/c0ob00564a-f1.gif) | ||
| Fig. 1 Radio-HPLC trace of crude material [18F]6a. | ||
Conclusions
In conclusion, we have demonstrated that the Staudinger ligation is a suitable strategy for prosthetic group labelling using [18F]2-fluoroethylazide. This catalyst-free ligation process benefits from short reaction times and very high radiochemical yields, a clear advantage for multi-step indirect 18F-labelling. Fluorous technology has been implemented for the fast and highly efficient purification of the prosthetic group [18F]2-fluoroethylazide and for the traceless Staudinger ligation itself. In the context of 18F-radiochemistry, this is the first example of fluorous detagging by indirect 18F-labelling.16 In recognition of the importance of the Staudinger ligation in the conjugation of biomolecules, we anticipate that this novel prosthetic group 18F-radiochemistry may find numerous applications in the context of peptide 18F-labelling as the new functionality installed upon ligation is the native amide bond.Experimental
General methods
1H NMRs were reported on Bruker DPX 200, DPX 400, AV 400 and AV 500 spectrometers, at a frequency of 200, 400 and 500 MHz respectively. 13C NMRs were recorded on Bruker AV 400 and AV 500 spectrometers at a frequency of 100 or 125 MHz respectively. Mass spectra (m/z) were obtained on a Bruker MicroTOF in Electrospray (ESI). Analytical thin layer chromatography (TLC) was performed on Merck Silica 60 F254 plates. Crude reaction mixtures were analysed by TLC and HPLC. HPLC analysis was performed with a Gilson 322 or Dionex Ultimate 3000 system, equipped with a NaI/PMT radiodetector (Raytest) and a UV-detector (Gilson). Radio-TLC was performed on Macherey-Nagel Polygram Silica Plates and eluted with EtOAc or 95% aq. MeCN. Analysis was performed with a plastic scintillator/PMT detector. FSPE separation was carried out using pre-assembled Waters Sep-Pak cartridges (Waters, Milford, MA) and FluoroFlash Silica gel (Fluorous Technologies Inc., Pittsburgh, PA). Pre-assembled Sep-Pak C18SPE cartridges (Waters, Milford, MA) were used in the same way.[18F]Fluoride was produced by the cyclotron of PETNET Solutions at Mont Vernon Hospital (UK) via the 18O(p,n)18F reaction and delivered as [18F]fluoride in [18O]water (1–2 GBq, 1–3 mL). This target solution was passed through a QMA anion exchange resin cartridge (20 mg, Waters). [18F]Fluoride adsorbed on the charged-resin was eluted into a reaction vial with a solution of Kryptofix 222 (15 mg) and K2CO3 (3 mg) in 1 mL acetonitrile–water (8:2). Excess water was removed under N2 stream at 100–110 °C, and the resulting complex was dried an additional 3 times by azeotropic distillation with 0.5 mL acetonitrile each under N2 stream. The resulting dry complex of K18F/Kryptofix 222 was further dissolved by anhydrous acetonitrile (2–4 mL) and dispensed into reaction vials containing the precursor for nucleophilic fluorination.
Selected experimental data are given below—full experimental procedures and spectroscopic data for all compounds in the paper are given in the supporting information.
General procedure for compounds 5a–d
(Diphenylphosphino)methanethiol (2) (416 mg, 1.8 mmol) was dissolved in DCM (20 mL), to which DCC (408 mg, 1.98 mmol), DMAP (1 crystal) and then carboxylic acid (1.8 mmol) were added. The reaction was stirred for 16 h at room temperature, during which time, a white precipitate formed. The reaction mixture was passed through Celite® and concentrated to dryness. The residue was purified by flash chromatography (40% EtOAc in hexane) to afford the desired product which was subsequently dissolved in toluene (5 mL) and DABCO (92 mg, 0.82 mmol) was added. The reaction mixture was stirred at 60 °C for 1 h, at which point it was cooled to room temperature and concentrated to dryness. The residue was purified by flash chromatography (50% EtOAc in hexane) to afford the desired products 5.General procedure for compounds 6a–d
Phosphinothioate 5 (0.05 mmol) was dissolved in a mixture of THF and water (3:1, 1 mL) and 2-fluoroethylazide (150 μL of a 0.33 M solution in THF, 0.05 mmol) was added. The reaction mixture was stirred at room temperature for 16 h then concentrated in vacuo. The residue was purified by flash chromatography (95:5 DCM–MeOH) to afford the desired products 6.
:1, 1 mL) and 2-fluoroethylazide (390 μL of a 0.33 M solution in THF, 0.13 mmol) was added. The reaction mixture was stirred at room temperature for 16 h then concentrated in vacuo. The residue was purified by FSPE to afford the desired product 6c in 27% yield.
Radiochemical procedures
2-[18F]fluoroethylazide [18F]1 was prepared as previously reported and purified by FSPE.11General procedure for 18F-labelled Staudinger ligation reactions
In a sealed reaction vial, the FSPE purified solution of 2-[18F]fluoroethylazide (20–100 MBq) in MeCN–H2O (7:3, 0.5 mL) was mixed with a thiophosphane 5 (1 mg) in either 300 mL THF–H2O (4:1) or 300 mL DMF–H2O (6:1) and heated for 15 min at 120 °C. Analysis by reverse-phase HPLC gave retention times that correlated to the cold reference compounds, with >95% conversion from [18F]2-fluoroethylazide to the product [18F]6 in each case.
Acknowledgements
We are grateful for financial support from the DIUS and Siemens Molecular Imaging (Project No TP/16636) (LC, RB) and from AstraZeneca (SB).References
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106 CrossRef; J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211 CrossRef CAS; E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS.
- For selected examples, see: E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 Search PubMed; M. J. Hangauer and C. R. Bertozzi, Angew. Chem., Int. Ed., 2008, 47, 2394 CrossRef CAS; E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS; G. Charron, M. M. Zhang, J. S. Yount, J. Wilson, A. S. Raghavan, E. Shamir and H. C. Hang, J. Am. Chem. Soc., 2009, 131, 4967 CrossRef CAS; P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821 CrossRef CAS; J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS.
- S. A. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501 CrossRef CAS.
- R. Schirrmacher, C. Wängler and E. Schirrmacher, Mini-Rev. Org. Chem., 2007, 4, 317 Search PubMed; L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853 CrossRef CAS; P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS.
- J. Marik and J. L. Sutcliffe, Tetrahedron Lett., 2006, 47, 6681 CrossRef CAS; M. Glaser and E. Årstad, Bioconjugate Chem., 2007, 18, 989 CrossRef CAS; G. Smith, M. Glaser, M. Perumal, Q.-D. Nguyen, B. Shan, E. Årstad and E. O. Aboagye, J. Med. Chem., 2008, 51, 8057 CrossRef CAS; M. Glaser, M. Solbakken, D. R. Turton, R. Pettitt, J. Barnett, J. Arukwe, H. Karlsen, A. Cuthbertson, S. J. Luthra and E. Arstad, Amino Acids, 2009, 37, 717 CrossRef CAS; M. Glaser and E. G. Robbins, J. Labelled Compd. Radiopharm., 2009, 52, 407 CrossRef CAS.
- B. L. Nilsson, L. K. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939 CrossRef CAS; E. Saxon, J. L. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141 CrossRef CAS; B. L. Nilsson, L. K. Kiessling and R. T. Raines, Org. Lett., 2001, 3, 9 CrossRef CAS.
- B. L. Nilsson, R. J. Hondal, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 5269.
- M. B. Soellner, B. L. Nilsson and R. T. Raines, J. Org. Chem., 2002, 67, 4993 CrossRef CAS; Y. He, R. J. Hinklin, J. Chang and L. K. Kiessling, Org. Lett., 2004, 6, 4479 CrossRef CAS.
- Deprotection was performed immediately prior to ligation with 1 as adducts 6 were found to be susceptible to oxidation.
- For selected reviews on fluorous synthesis see: W. Zhang, Tetrahedron, 2003, 59, 4475 Search PubMed; W. Zhang, Chem. Rev., 2004, 104, 2531 CrossRef CAS; W. Zhang, Chem. Rev., 2009, 109, 749 CrossRef CAS.
- R. Bejot, T. Fowler, L. Carroll, S. Boldon, J. E. Moore, J. Declerck and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 586 CrossRef CAS.
- E. Blom, F. Karimi and B. Långström, J. Label. Compd. Radiopharm., 2010, 53, 24 CAS.
- For an example of fluorous electrophilic fluorodetagging: S. Boldon, J. E. Moore and V. Gouverneur, Chem. Commun., 2008, 3622 Search PubMed.
- For a fluorous Staudinger reaction: C. W. Lindsley, Z. Zhao, R. C. Newton, W. H. Leister and K. A. Strauss, Tetrahedron Lett., 2002, 43, 4467 Search PubMed; D. P. Curran, X. Wang and Q. Zheng, J. Org. Chem., 2005, 70, 3716 CrossRef CAS.
- C. Mamat, A. Flemming, M. Köckerling, J. Steinbach and F. R. Wuest, Synthesis, 2009, 3311 CrossRef CAS.
- During the preparation of this manuscript, the Staudinger Ligation has been reported for the radiosynthesis of [18F]GABAA receptor binding 4-quinolones: A Gaeta, J. Woodcraft, S. Plant, J. Goggi, P. Jones, M. Battle, W. Trigg, S. Luthra and M. Glaser, Bioorg. Med. Chem. Lett., 2010, 20, 4649 Search PubMed.
Footnotes |
| † This publication is part of the web themed issue on fluorine chemistry. |
| ‡ Electronic supplementary information (ESI) available: HPLC data of radiolabelling experiments. See DOI: 10.1039/c0ob00564a |
| This journal is © The Royal Society of Chemistry 2011 |