Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates†‡§
Armin
Thalhammer
a,
Jasmin
Mecinović
b,
Christoph
Loenarz
a,
Anthony
Tumber
c,
Nathan R.
Rose
a,
Tom D.
Heightman
c and
Christopher J.
Schofield
a
aDepartment of Chemistry and the Oxford Centre for Integrative Systems Biology, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, United Kingdom. E-mail: Christopher.schofield@chem.ox.ac.uk; Fax: +44 1865 285002; Tel: +44 1865 275625
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford St, Cambridge, MA 02138, USA
cStructural Genomics Consortium, University of Oxford, Headington, OX3 7DQ, United Kingdom
First published on 15th November 2010
Abstract
Based on structural analysis of the human 2-oxoglutarate (2OG) dependent JMJD2 histoneNε-methyl lysyl demethylase family, 3-substituted pyridine 2,4-dicarboxylic acids were identified as potential inhibitors with possible selectivity over other human 2OG oxygenases. Microwave-assisted palladium-catalysed cross coupling methodology was developed to install a diverse set of substituents on the sterically demanding C-3 position of a pyridine 2,4-dicarboxylate scaffold. The subsequently prepared di-acids were tested for in vitro inhibition of the histone demethylase JMJD2E and another human 2OG oxygenase, prolyl-hydroxylase domain isoform 2 (PHD2, EGLN1). A subset of substitution patterns yielded inhibitors with selectivity for JMJD2E over PHD2, demonstrating that structure-based inhibitor design can enable selective inhibition of histone demethylases over related human 2OG oxygenases.
Introduction
Covalent modifications to histone proteins play important roles in cell differentiation, embryonic development and cancer. The activities of enzymes that catalyse such modifications can regulate gene expression and have also been associated with diseases, including cancer (for reviews, see1–3). Histone deacetylases have been successfully targeted using small-molecule histone deacetylase (HDAC) inhibitors such as suberoylanilide hydroxamic acid4 (SAHA) and the natural product Romidepsin,5 which are approved for use in clinical cancer treatment. Similar approaches might be viable for the modulation of histone methyl transferase and histone demethylase activities; Indeed, some histone methylation patterns have been observed to correlate with disease pathology (reviewed in6–8).In humans, there are five identified subfamilies of 2-oxoglutarate (2OG) dependent histone demethylases and several of these enzymes are of clinical relevance.9 The best characterised subfamily of the human 2OG dependent histone demethylases are the Jumonji domain containing 2 (JMJD2) enzymes, the genes for four of which are expressed (JMJD2A-D), whereas JMJD2E and JMJD2F are annotated as pseudogenes.10 The JMJD2 enzymes appear to be selective for demethylation of histone H3 at lysine 9 (H3K9) and, in the case of JMJD2A-C, additionally for lysine 36 of histone H3 (H3K36).9JMJD2 histone lysyl demethylases differ in their preference for the methylation state of their substrates, catalysing the selective demethylation of tri- and di- and, to a lesser extent, the mono-Nε-methylated forms of their lysine substrates, producing formaldehyde as a byproduct11 (Scheme 1A).
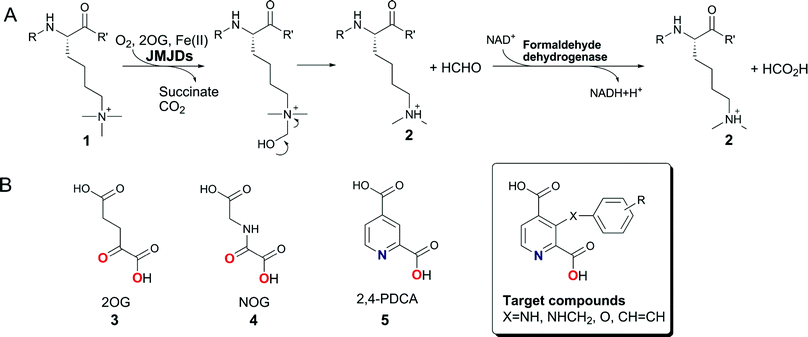 | ||
| Scheme 1 A. The Nε-methyl lysine demethylation reaction catalysed by the 2OG-dependent JMJD2 histone demethylases, which preferentially act on tri- and di-Nε-methylated lysines. Demethylation of Nε-trimethyllysyl residues (1) proceeds viahydroxylation to give an unstable hemiaminal intermediate, which collapses spontaneously to release the demethylation product 2. The formation of the by-product formaldehyde can be measured spectrophotometrically using formaldehyde dehydrogenase and NAD+ in a coupled enzymatic assay. B. Structures of the 2OG cosubstrate (3), its analogues NOG (4) and 2,4-PDCA (5), and the targeted C-3 substituted 2,4-PDCA derivatives. The ligand atoms involved in chelation of the ferrous iron are shown in red and blue. | ||
There is significant interest in the development of selective histone demethylase inhibitors, both for biomedicinal studies and as functional probes aimed at defining the roles of these enzymes in epigenetic regulation.12 The design of inhibitor scaffolds with selectivity for inhibition of individual histone demethylases or subfamilies of histone demethylases is hampered by the five 2OG dependent histone demethylase subfamilies being members of the wider 2OG oxygenase family, which has ∼60–80 human members with diverse biochemical roles in oxygen sensing, fat mass regulation, mRNA splicing, DNA demethylation, and 5-methylcytosine hydroxylation.13,14
JMJD2E is closely related to the catalytic domain of the other JMJD2 enzymes (∼65% sequence identity with JMJD2A over the catalytic domain) and can be prepared in an active form, which is amenable to kinetic analysis.15 The availability of recombinant JMJD2E enabled the identification of several “template” inhibitors, most of which work by Fe(II) chelation at the active site and compete with the 2OG cosubstrate for binding.15 Recently, Hamada et al.16 reported that hydroxamate derivatives were JMJD2A inhibitors; In cell-penetrating form, some of the compounds were shown to inhibit cancer cell growth when used in combination with an inhibitor of the pyridoxal phosphate lysine-specific class of histone demethylases. Another type of JMJD2 subfamily inhibitors works by disrupting a Zn(II) binding site separate from the active site.17 Transition metal ions also display differential inhibition selectivities for JMJD2A, JMJD2E and the hypoxia-inducible prolyl hydroxylase domain 2 (PHD2, EGLN1), a somewhat structurally different 2OG oxygenase that is involved in the adaptation of cells to hypoxia.18 In addition to N-oxalyl amino acid derivatives,12,15,19 a previous study identified pyridine dicarboxylates as potential JMJD2 inhibitors;15 however, these pyridine dicarboxylates are known to inhibit all other studied members of the 2OG oxygenase family.15 Interestingly, potent inhibition of JMJD2 enzymes has been observed with 2,4-pyridine dicarboxylic acid (2,4-PDCA, Scheme 1A), but not with its other regioisomers that were also tested, suggesting that differential substitution of 2,4-PDCA derivatives may help to enable the development of JMJD2-selective inhibitors.15 Crystallographic analyses of JMJD2A in complex with 2,4-PDCA have shown that it binds in a similar manner to the 2OG cosubstrate,15 being positioned to bind to the active site metal ion in a bidentate manner, while the C-4 acid is positioned for electrostatic interactions with the sidechain of Lys-206, as also occurs with the C-5 carboxylate of 2OG (Fig. 1C,D). One advantage of a substituted 2,4-PDCA scaffold is that its methyl and ethyl esters have been shown to be cell-penetrating and to inhibit histone demethylase activity in cells,20,21 thus suggesting that in vivo applications might be possible.
![Crystallographic analyses on JMJD2A and PHD2. A. View from a crystal structure of JMJD2A in complex with NiII (substituting for FeII) and the inhibitor 2,4-PDCA (derived from PDB ID2VD715). B. Active site of JMJD2A showing chelation of the active site metal (green) by the conserved iron-binding Hx[D/E]…H motif (His188, Glu190 and His276). Selected active site residues are shown. C. View from a crystal structure of the prolyl hydroxylase PHD2 in complex with MnII, a fragment of the hypoxia inducible factor 1α peptide substrate (not shown), and the 2OG cosubstrate analogue N-oxalyl glycine (NOG), a relatively non-specific 2OG oxygenase inhibitor (PDB ID3HQR22). D. Closeup view of the PHD2 active site showing chelation of MnII (green) by the conserved iron-binding Hx[D/E]…H motif (His313, Asp315 and His374).](/image/article/2011/OB/c0ob00592d/c0ob00592d-f1.gif) | ||
| Fig. 1 Crystallographic analyses on JMJD2A and PHD2. A. View from a crystal structure of JMJD2A in complex with NiII (substituting for FeII) and the inhibitor 2,4-PDCA (derived from PDB ID2VD715). B. Active site of JMJD2A showing chelation of the active site metal (green) by the conserved iron-binding Hx[D/E]…H motif (His188, Glu190 and His276). Selected active site residues are shown. C. View from a crystal structure of the prolyl hydroxylase PHD2 in complex with MnII, a fragment of the hypoxia inducible factor 1α peptide substrate (not shown), and the 2OG cosubstrate analogue N-oxalyl glycine (NOG), a relatively non-specific 2OG oxygenase inhibitor (PDB ID3HQR22). D. Closeup view of the PHD2 active site showing chelation of MnII (green) by the conserved iron-binding Hx[D/E]…H motif (His313, Asp315 and His374). | ||
Here we report the preparation of C-3 substituted 2,4-PDCA derivatives, and their evaluation as JMJD2 inhibitors. The results demonstrate how structure-guided substitution of a generic inhibitor template can be used to develop selective inhibitors of JMJD2E over PHD2.
Results and Discussion
Synthesis of 3-bromo-2,4-PDCA dimethyl ester
To obtain insights as to which inhibitor scaffold modifications are most likely to achieve selectivity between different 2OG oxygenase families, we compared reported crystal structures of human 2OG oxygenases. Amongst others, co-crystal structures of complexes with inhibitors have been reported for JMJD2A (JMJD2A·2,4-PDCA, PDB ID2VD715) and for PHD2 in complex with a fragment of its hypoxia inducible factor (HIF) peptide substrate (PHD2·NOG·HIF-1α558–574, PDB ID3HQR22). These analyses suggested that substitution of 2,4-PDCA at the C-3 position, rather than the C-5 or C-6 positions, would enable functional groups to form additional interactions with the substrate binding pocket of the JMJD2s, and could potentially abolish binding to the more restricted 2OG binding site of PHD2 (Fig. 1B and 1D). We therefore targeted 2,4-PDCA derivatives substituted at C-3 with an additional phenyl ring that could bear a diverse set of substituents (Scheme 1B).The 3-bromopyridine 14 was considered to be a potential intermediate for the preparation of several C-3 substituted 2,4-PDCA derivatives (Scheme 2). Preparation of 14 was initially attempted via3-hydroxy-2,4-PDCA 9, which could also serve as a parent compound for the preparation of O-modified derivatives itself. 3-Hydroxypyridine (6) was O-alkylated with tetrahydropyran-2-ol under Mitsunobu conditions23 or with methoxymethyl chloride to furnish derivatives 7 and 8 as reported.24 However, attempted directed metallation-carboxylation25 by treatment with n-BuLi and quenching with CO2(s), after hydrolytic workup, only gave 4-carboxy-3-hydroxypyridine in low yields. Conditions employing either s-BuLi or t-BuLi as a base resulted in complex product mixtures.
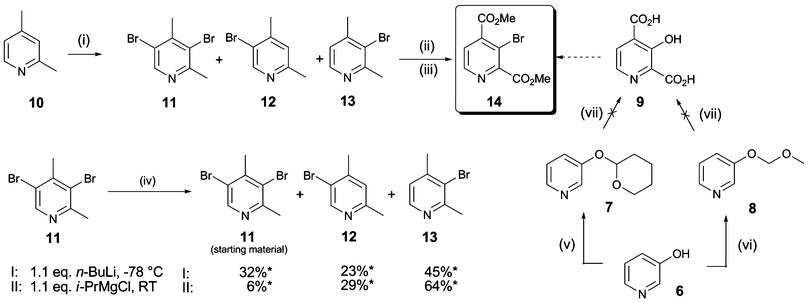 | ||
| Scheme 2 Synthesis of 3-bromopyridine 14. Reagents and conditions: (i) Br2 (0.9 eq.), 20% oleum, 165 °C, 24 h, 11 (12%), 12 (22%), 13 (35%); (ii) KMnO4 (5 eq.), NaOH (0.7 eq.), H2O, 110 °C, 3 h; (iii) cat. H2SO4, MeOH, reflux, 24 h, 53%; (iv) n-BuLi (1.1 eq.), THF, −78 °C, 1 h, or i-PrMgCl (1.1 eq.), THF, RT; then MeOH workup; (v) tetrahydropyran-2-ol (1.1 eq.), PPh3 (1.1 eq.), diisopropyl azodicarboxylate (1.1 eq.), RT, 2 h, 46%;23(vi) methoxymethyl chloride (1.05 eq.), KOtBu (1.1 eq.), DMF–CH3CN, RT, 1 h;26 (vii) base (1.5 eq.), then excess CO2(s); base (1.5 eq.), then excess CO2(s); then HCl/dioxane; *Ratios as determined by 1H NMR. | ||
In an alternative approach, electrophilic bromination of 2,4-dimethylpyridine 10 in 20% oleum for 24 h at 165 °C as described27 gave a mixture of 3,5-dibromo-2,4-dimethylpyridine 11 (12%), 5-bromo-2,4-dimethylpyridine 12 (22%), and unreacted starting material, in addition to the desired 3-bromo-2,4-dimethylpyridine 13 (35% yield after chromatography). When a larger excess of bromine was used (2 eq.), the reaction went to completion; however, the yield of the dibromopyridine 11 increased at the expense of 13. When the oleum concentration was decreased to 10% (w/v), in an attempt to enhance the selectivity of bromination, no conversion was observed. To improve the overall yield of the desired isomer 13, the regioselective debromination of 11 was investigated. Treatment of the dibrominated compound 11 with n-BuLi, or i-PrMgCl, followed by protonation of the metallated intermediates, gave mixtures of isomeric mono- (12 and 13) and dibromo (11) 2,4-dimethylpyridines. 1H NMR analysis of the crude products indicated a preference for the formation of the desired 3-bromo substituted isomer, suggesting that the C-5 position of 2,4-dimethylpyridine is sterically more accessible to halogen-metal exchange. By this procedure, 11 was recycled to increase the overall yield of 13. Permanganate oxidation and esterification then afforded the protected intermediate scaffold 14.
Transition-metal catalysed coupling reactions
We then investigated the formation of C–C bonds at the C-3 position of 2,4-PDCA (Schemes 3 and 4). Application of cross-coupling methodology with intermediate 14 as a substrate was found to require optimisation, likely because of steric hindrance at C-3 and because both 14 and the coupling products are prone to base-catalysed hydrolysis and thermal decarboxylation.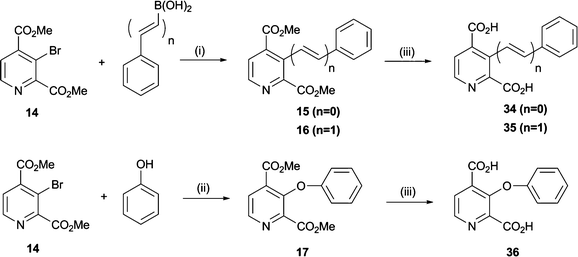 | ||
| Scheme 3 Synthesis of pyridine dicarboxylate derivatives 34–36. Reagents and conditions: (i) Pd(OAc)2 (10 mol%), PPh3 (0.2 eq.), Cs2CO3 (1.1 eq.), DMF, 70 °C, 3–5 h, 60–75%; (ii) CuI (0.1 eq.), N-(n-butyl)imidazole (0.5 eq.), Cs2CO3 (2 eq.), toluene, MW, 140 °C, 1 h, 44%; (iii) NaOH, MeOH, H2O, 6 h-overnight, 49–75%. | ||
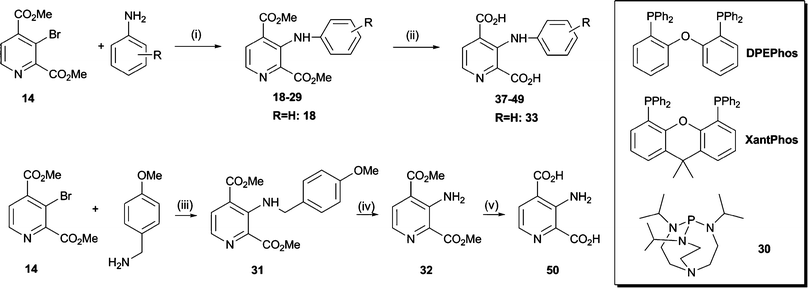 | ||
| Scheme 4 Synthesis of pyridine dicarboxylate derivatives 37–50. Reagents and conditions: (i) Pd2dba3 (2 mol%), ligand L (6 mol%), base (1.4 eq.), toluene, MW, 110–150 °C; L = PPh3 or BINAP or 30: <5% conversion; L = DPEPhos: 11% yield; L = XantPhos: 77% yield; (ii) NaOH, MeOH–H2O, RT; (iii) Pd2dba3 (2 mol%), XantPhos (6 mol%), Cs2CO3 (1.4 eq.), toluene, MW, 110 °C, 12 h, 69%; (iv) CF3CO2H, CH2Cl2, 0 °C to RT, 89%; (v) NaOH, MeOH–H2O, 2 h, RT, 67%. | ||
Carbon–carbon bond formation28 with 14 was achieved in 75% yield using Suzuki–Miyaura couplings with phenyl boronic acid/anhydride, enabling the direct incorporation of a phenyl ring at the C-3 position of 2,4-PDCA (15) (Scheme 3). Similarly, the styryl derivative (16) was obtained from styryl boronic acid in 60% yield. Attempts to prepare 16 by Heck coupling of 14 with styrene were unsuccessful, despite investigation of common catalyst and solvent systems.
Efforts were then directed to identify a suitable catalyst and solvent system for the introduction of oxygen and nitrogen nucleophiles at the C-3 position of 14, initially using phenol and aniline as model substrates. Cu(II)-catalysed Ullmann-type cross-coupling reactions, which are commonly employed for diarylether formation, usually require high reaction temperatures and long reaction times, especially on sterically demanding scaffolds.29 As might be expected, treatment of 14 with an excess of phenol and CuO at 200 °C for 24 h resulted in decomposition of starting material. Similarly, a catalyst system comprising Pd(OAc)2, Cs2CO3 and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos) was ineffective. In contrast, the use of N-(n-butyl)imidazole and CuI under mild phase-transfer conditions30 with microwave heating enabled formation of the diarylether 17 in moderate yield.
Couplings with aromatic amines using Buchwald–Hartwig conditions31 were then optimised on a model system comprising 14, aniline, cat. Pd(II) and a set of phosphine ligands (Scheme 4). With XantPhos, 18 was obtained in 77% isolated yield, whereas bis(2-diphenylphosphinophenyl)ether (DPEPhos) catalysis gave only 11% yield. No significant product formation was observed when using either (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene (BINAP), triphenylphosphine or the bicyclo[3.3.3]undecane derivative 30 as catalysts. These results are consistent with the proposal that the ‘bite angle’ (the phosphorus-metal-phosphorus angle) of the phosphine ligand plays a role in enabling efficient catalysis.32 Amongst a number of solvent and base combinations investigated, toluene and Cs2CO3 proved to be a suitable combination; significant hydrolysis of the starting material was not observed under these conditions. Aromatic amines with a range of functional groups, including nitro, carboxylic acid, methoxy and fluorine moieties, were used as coupling partners under these conditions. In general, reactions proceeded faster and more smoothly with aryl amines containing an ortho-substituent or an electron-donating para-substituent than those bearing an electron-withdrawing para-substituent. In the latter case, coupling yields were considerably lower and longer reaction times as well as elevated reaction temperatures were required.
We then aimed to obtain the 3-amino-2,4-PDCA dimethyl ester 32 in order to evaluate the electronic effect of an amino group at the pyridine C-3 position on inhibitor potency, independently of steric effects introduced by more bulky substituents. Because “classical” Buchwald–Hartwig couplings are limited to the preparation of secondary and tertiary amines, ammonia surrogates have been developed which also allow the preparation of primary amines using transition-metal catalysis. For example, anilines can be prepared by palladium-catalysed amination of aryl halides with benzophenone imine to afford intermediate benzophenone imines, which are readily hydrolysed to the desired anilines.33 However, attempted coupling of 3-bromo-2,4-PDCA 14 with benzophenone imine was inefficient, possibly due to steric limitations. Alternatively, cross-couplings with benzyl amine followed by hydrogenolysis have been used to install primary amino groups, for example in the synthesis of 3-aminoestrone.34 We therefore envisaged that the related p-methoxybenzylamine could serve as ammonia equivalent in a Buchwald–Hartwig type cross-coupling reaction and could be readily removed by acid-mediated cleavage. Indeed, reaction of 3-bromo-2,4-PDCA 14 with p-methoxybenzylamine afforded the targeted amino derivative 31 in 69% yield; subsequent deprotection with CF3CO2H in CH2Cl2 gave ester 32.
The methyl esters thus obtained could further be employed to obtain cell-penetrable inhibitors.20,21 For in vitro experiments, the desired dicarboxylic acids 33–50 were prepared by alkaline hydrolysis of the corresponding dimethyl esters and typically did not require further purification, being >95% pure by HPLC and NMR analyses.
Biological results
An initial series of compounds was prepared to investigate the preferred atoms at the C-3 position of 2,4-PDCA. The JMJD2E inhibitory activities of compounds 33–50 against JMJD2E were measured as IC50 values in a formaldehyde dehydrogenase (FDH) coupled, spectrophotometric turnover assay, which measures production of formaldehyde (Scheme 1A, Table 1 and ESI§).15 Compounds were then counterscreened against FDH to confirm genuine JMJD2E inhibition. To investigate potential selective inhibition of JMJD2s by some of the synthesised 3-substituted 2,4-PDCA derivatives, they were tested for inhibition of PHD2. Residual activities of PHD2 were determined at 400 μM inhibitor concentration, using a fluorescence-based assay.35,36|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | Template | R | JMJD2E IC50/μMa | JMJD2E Residual Activity (%)a | PHD2 Residual Activity (%)b | FDH Residual Activity (%)c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Determined by FDH-coupled assay; enzyme concentration 400 nM; residual activity reported at 200 μM compound concentration. b Determined by an HTRF-based assay; enzyme concentration 20 nM; residual activity reported at 400 μM compound concentration. c Enzyme concentration 50 μM; spectrophotometric assay; residual activity reported at 200 μM compound concentration. d Compound was fluorescent at the tested concentration. e Stock solutions prepared at 1 mM concentration because of limited solubility. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 5 | A | H | 0.44 | 0% | 22% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 33 | A | Br | n.d. | 67% | 100% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 34 | A | Ph | n.d. | 84% | 100% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 35 | A | (E)-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh CHPh |
66 | 25% | 85% | 87% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 36 | A | OPh | n.d. | 48% | 88% | 91% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 37 | B | NHPh | 19 | 0% | 100% | 83% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 38 | B | NH-p-C6H4-OMe | 164 | 6% | 100% | 78% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 39 | B | NH-p-C6H4-NO2 | 95 | 0% | 100% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 40 | B | NH-p-C6H4-CO2H | 66 | 13% | 0% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 41 | B | NH-o,p-C6H3-diF | 48 | 12% | 100% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 42 | B | NH-o-C6H4-SMe | 48 | 5% | 100% | 80% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 43 | B | NH-o-C6H4-CO2H | 166 | 39% | 100% | 73% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 44 | B | NH-o-C6H4-NO2 | 56 | 6% | 100% | 59% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 45 | B | NH-o-C6H4-OMe | 19 | 2% | 100% | 76% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 46 | B | NH-o-C6H4-Me | 41 | 3% | 100% | 79% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 47 | B | NH-o-C6H4–F | 2.5 | d | 100% | d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 48 | A | NH-Naphthyl | 26 | 0% | 100% | 59% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 49 | A | NH-CH2-p-C6H4-OMe | 41 | 29% | 71% | 100% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | 50 e | A | NH2 | 0.11 | d | 23% | d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
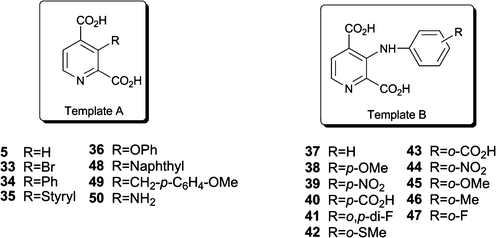
Replacement of hydrogen in the 3-position of 2,4-PDCA by bromine or phenyl moieties resulted in a significant reduction of potency against JMJD2E (IC50 > 100 μM) and also of potency against PHD2, possibly due to steric effects on the geometry of the carboxylate groups, causing sub-optimal Fe(II) coordination, and/or binding to the arginyl (PHD2) and lysyl (JMJD2A) residues that bind the C-4 carboxylate of 2,4-PDCA (Table 1, entries 1–3, Fig. 1B and 1D). In support of this proposal was the observation that upon installation of an ethylene linker between the two aromatic rings (styryl derivative 35), some recovery of inhibitory potency was observed (Table 1, entry 4).
Interestingly, 3-amino-2,4-PDCA 50 was a highly active JMJD2E inhibitor (IC50 ∼100 nM), although it displayed little selectivity for JMJD2E over PHD2 (Table 1, entry 19). Although 50 was fluorescent at the initial assay concentration tested, it gave an acceptable dose-response curve. These results reveal that the presence of a C-3 amino group promotes the binding of the 2,4-PDCA template to JMJD2A (and PHD2). It is possible that the additional amino group enhances potency, at least in part by enabling pre-organisation of the two carboxylate groups into coplanarity, thus favouring a conformation similar to that observed in the co-crystal structure of 2,4-PDCA with JMJD2A (Fig. 1A and 1B). Addition of a phenyl ring to the exocyclic amino group of 3-amino-2,4-PDCA (37) ablated inhibition of PHD2 within the limits of detection (100% residual activity at 400 μM), whilst retaining significant activity against JMDJ2E (IC50 19 μM). In contrast, the phenoxy-derivative 36 was inactive against both JMJD2E and PHD2 (Table 1, entries 5–6).
Having established that selectivity for JMJD2E over PHD2 can be enhanced by C-3 substitution of 2,4-PDCA, efforts were directed at increasing inhibitory potency against JMDJ2E whilst retaining selectivity. Thus, further analogues (38–47) were designed and prepared to evaluate the effect of substituents on the 3-aminophenyl ring. The para-substituted compounds 38–41 were less potent than the unsubstituted system 37 (Table 1, entries 6–10). In contrast, the ortho-substituted derivatives 41–47 displayed a broad range of inhibitory activities against JMJD2E (Table 1, entries 10–16). Compounds bearing methoxy- (45) and fluoro- (47) substituents were significantly more potent than those with carboxylate (43), nitro (44) or methyl (46) groups. These results suggest that a combination of steric and electronic effects determine potency in the 3-substituted 2,4-PDCA series. One of the ortho-substituted 3-phenyl derivatives (40) showed substantial inhibition of PHD2 at 400 μM (Table 1, entry 9). No significant FDH inhibition was observed for the potent compounds at concentrations close to their IC50 value against JMJD2E (Table 1), confirming genuine JMJD2E inhibition. Therefore, among others, compounds 37, 45, 46, 48, and, in particular, 47 and 50, represent selective inhibitors of the JMJD2E histone demethylase over the related human 2OG dependent oxygenase PHD2.
Conclusions
Based on structural analyses, we identified a series of C-3 substituted 2,4-PDCA derivatives as inhibitors of the JMJD2 subfamily of histone demethylases. Synthetic methods employing Pd-catalysed couplings were developed and applied to the preparation of C-3 substituted 2,4-PDCA compounds derived from dimethyl 3-bromopyridine-2,4-dicarboxylate 14. The results demonstrate that compounds such as 37, 45 and 47 are potent JMJD2E inhibitors. Importantly, these compounds do not inhibit PHD2, as predicted on the basis of structural analyses, and may be useful in their esterified forms as inhibitors for the dissection of the biological roles of different 2OG oxygenase subfamilies.19 Thus, our work demonstrates that structural analyses of the catalytic site pocket of human 2OG oxygenases can be used both to enhance potency to the target demethylases and to block binding to other human 2OG oxygenases.Experimental
Representative synthetic procedures
CH), 7.23–7.30 (1 H, t, J 8.0 Hz, ArHpara), 7.34 (2 H, t, J 8.0 Hz, ArHmeta), 7.46 (2 H, d, J 7.0 Hz, ArHortho), 7.64 (1 H, d, J 16.5 Hz, CHCH), 7.71 (1 H, d, J 5.0 Hz, pyH) and 8.65 (1 H, d, J 5.0 Hz, pyH); δC (100 MHz; CDCl3) 52.8, 52.8, 123.5, 124.6, 126.7, 128.4, 128.7, 133.0, 135.1, 136.5, 139.0, 147.9, 149.8, 166.5 and 166.7; m/z (ESI+) 320.0893 (M + Na+. C17H15NNaO4 requires 320.0893).
CH), 7.30 (1 H, t, J 7.5 Hz, ArHpara), 7.37 (2 H, t, J 7.5 Hz, ArHmeta), 7.51 (2 H, d, J 7.5 Hz, ArHortho), 7.69 (1 H, d, J 16.5 Hz, CHCH), 7.85 (1 H, d, J 5.0 Hz, pyH), 8.62 (1 H, d, J 5.0 Hz, pyH); δC (100 MHz; MeOD) 123.7, 125.0, 126.8, 128.4, 128.8, 132.5, 135.2, 137.2, 141.6, 147.5, 150.5, 168.2, 168.5; m/z (ESI-) 268.0616 (M − H+. C15H10NO4 requires 268.0615).
Acknowledgements
We thank Thomas Helleday for encouragement, John M. Brown for helpful discussions, and the Wellcome Trust, the Biotechnology and Biological Sciences Research Council and Cancer Research UK for financial support.References
- Y. Shi, Nat. Rev. Genet., 2007, 8, 829–833 CrossRef CAS.
- P. A. C. Cloos, J. Christensen, K. Agger and K. Helin, Genes Dev., 2008, 22, 1115–1140 CrossRef CAS.
- B. R. Keppler and T. K. Archer, Expert Opin. Ther. Targets, 2008, 12, 1301–1312 CrossRef CAS.
- P. A. Marks, Oncogene, 2007, 26, 1351–1356 CrossRef CAS.
- J. Tan, S. Cang, Y. Ma, R. Petrillo and D. Liu, J. Hematol. Oncol., 3 Search PubMed.
- S. Ng, W. Yue, U. Oppermann and R. Klose, Cell. Mol. Life Sci., 2009, 66, 407–422 CrossRef CAS.
- N. Mosammaparast and Y. Shi, Annu. Rev. Biochem., 2010, 79, 155–179 CrossRef CAS.
- R. J. Klose and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2007, 8, 307–318 CrossRef CAS.
- R. J. Klose, E. M. Kallin and Y. Zhang, Nat. Rev. Genet., 2006, 7, 715–727 CrossRef CAS.
- M. Katoh and M. Katoh, Int. J. Oncol., 2004, 24, 1623–1628 CAS.
- R. J. Hopkinson, R. B. Hamed, N. R. Rose, T. D. Claridge and C. J. Schofield, ChemBioChem, 2010, 11, 506–510 CrossRef CAS.
- N. R. Rose, E. C. Woon, G. L. Kingham, O. N. King, J. Mecinovic, I. J. Clifton, S. S. Ng, J. Talib-Hardy, U. Oppermann, M. A. McDonough and C. J. Schofield, J. Med. Chem., 2010, 53, 1810–1818 CrossRef CAS.
- C. Loenarz and C. J. Schofield, Nat. Chem. Biol., 2008, 4, 152–156 CrossRef CAS.
- C. Loenarz and C. J. Schofield, Chem. Biol., 2009, 16, 580–583 CrossRef CAS.
- N. R. Rose, S. S. Ng, J. Mecinovic, B. M. Lienard, S. H. Bello, Z. Sun, M. A. McDonough, U. Oppermann and C. J. Schofield, J. Med. Chem., 2008, 51, 7053–7056 CrossRef CAS.
- S. Hamada, T. Suzuki, K. Mino, K. Koseki, F. Oehme, I. Flamme, H. Ozasa, Y. Itoh, D. Ogasawara, H. Komaarashi, A. Kato, H. Tsumoto, H. Nakagawa, M. Hasegawa, R. Sasaki, T. Mizukami and N. Miyata, J. Med. Chem., 2010, 53, 5629–5638 CrossRef CAS.
- R. Sekirnik, N. R. Rose, A. Thalhammer, P. T. Seden, J. Mecinović and C. J. Schofield, Chem. Commun., 2009, 6376–6378 RSC.
- R. Sekirnik, N. R. Rose, J. Mecinovic and C. J. Schofield, Metallomics, 2010, 2, 397–399 RSC.
- S. Hamada, T.-D. Kim, T. Suzuki, Y. Itoh, H. Tsumoto, H. Nakagawa, R. Janknecht and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 2852–2855 CrossRef CAS.
- G. Tschank, D. G. Brocks, L. Engelbart, J. Mohr, E. Baader, V. Gunzler and H. Hanauske-Abel, Biochem. J., 1991, 275, 469–476 CAS.
- M. M. Mackeen, H. B. Kramer, K.-H. Chang, M. L. Coleman, R. J. Hopkinson, C. J. Schofield and B. M. Kessler, J. Proteome Res., 2010, 9, 4082–4092 CrossRef CAS.
- R. Chowdhury, M. A. McDonough, J. Mecinovic, C. Loenarz, E. Flashman, K. S. Hewitson, C. Domene and C. J. Schofield, Structure, 2009, 17, 981–989 CrossRef CAS.
- R. Azzouz, L. Bischoff, M.-H. Fouquet and F. Marsais, Synlett, 2005, 2808–2810 CAS.
- M. R. Winkle and R. C. Ronald, J. Org. Chem., 1982, 47, 2101–2108 CrossRef CAS.
- R. Azzouz, L. Bischoff, C. Fruit and F. Marsais, Synlett, 2006, 1908–1912 CAS.
- F. Le Strat, D. C. Harrowven and J. Maddaluno, J. Org. Chem., 2005, 70, 489–498 CrossRef CAS.
- A. D. Dunn and S. Guillermic, Z. Chem., 1988, 28, 59–60 CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS.
- T. Schareina, A. Zapf, A. Cotte, N. Muller and M. Beller, Tetrahedron Lett., 2008, 49, 1851–1855 CrossRef CAS.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS.
- J. P. Wolfe, J. Åhman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS.
- U. Schön, J. Messinger, M. Buchholz, U. Reinecker, H. Thole, M. K. S. Prabhu and A. Konda, Tetrahedron Lett., 2005, 46, 7111–7115 CrossRef.
- J. H. Dao, R. J. M. Kurzeja, J. M. Morachis, H. Veith, J. Lewis, V. Yu, C. M. Tegley and P. Tagari, Anal. Biochem., 2009, 384, 213–223 CrossRef CAS.
- C. Loenarz, J. Mecinović, R. Chowdhury, L. A. McNeill, E. Flashman and C. J. Schofield, Angew. Chem., Int. Ed., 2009, 48, 1784–1787 CrossRef CAS.
Footnotes |
| † We dedicate this paper to Robin G. Procter in the year of his retirement with thanks for many years of expert technical assistance in mass spectrometry. |
| ‡ This paper is part of an Organic & Biomolecular Chemistry web theme issue on chemical biology. |
| § Electronic supplementary information (ESI) available: Additional assay protocols, detailed inhibition data, and synthetic procedures for all compounds. See DOI: 10.1039/c0ob00592d |
| This journal is © The Royal Society of Chemistry 2011 |