Synthesis of dysideaproline E using organocatalysis†
Ernest
Owusu-Ansah
a,
Amanda C.
Durow
a,
John R.
Harding
b,
Angela C.
Jordan
b,
Susan J.
O'Connell
a and
Christine L.
Willis
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, UK BS8 1TS. E-mail: chris.willis@bristol.ac.uk; Tel: 0117 9287660
bAstraZeneca, Mereside Alderley Park, Macclesfield, Cheshire, UK SK10 4TG
First published on 15th November 2010
Abstract
(S)-4,4-Dichloro-3-methylbutanoic acid was prepared in 51% overall yield from commercially available starting materials using an organocatalytic transfer hydrogenation to 4,4-dichloro-3-methylbut-2-enal in the key step. The (S)-dichloro acid was used as an intermediate in the first total synthesis of dysideaproline E and a diastereomer confirming the structure of the natural product.
Introduction
An intriguing feature of many biologically active natural products is the presence of halogens. In the majority of cases these are located at sites in accord with biohalogenation occurring via electrophilic species, and haloperoxidases which catalyze such reactions have been widely studied.1 However an interesting family of chlorinated natural products exist in which the halogens are located at aliphatic positions remote from activating groups. The mechanism of biohalogenation via a remarkable direct chlorination of the pro-Rmethyl group of leucine has been a topic of much recent interest.2 Examples of such compounds are six related chlorinated natural products, dysideaprolines A to F (1–6) isolated from extracts of Dysidea sp (NCI 1517) collected in the Philippines (Fig. 1).3 The peptide structures (1–6) were determined by extensive spectroscopic methods and with the exception of dysideaproline D, 4, all contain the 4,4-dichloro-3-methylbutanoic acid 8 moiety. This was assigned the S configuration by analogy with other chlorinated natural products isolated from Dysidea sp.4Degradation studies on dysideaproline A, 1, revealed the S-configuration for C-13 but C-5 was not assigned. Further degradation studies on dysideproline E, 5, and Marfey analysis indicted that it is assembled on an N-methyl-D-leucine core i.e. C-5 has the R-configuration.3 Interestingly X-ray studies have revealed that sintokamide A (Fig. 1), a further polychlorinated natural product isolated from Dysidea sp., contains both D-trichloroleucine and L-dichloroleucine derived fragments and is an inhibitor of the androgen receptor in prostate cancer.5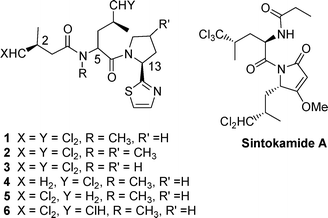 | ||
| Fig. 1 Examples of chlorinated natural products. | ||
Since these polychlorinated natural products are produced in only small quantities the development of methods for their synthesis is an important goal for structure confirmation and to provide material for full biological assessment. Herein we describe a practical and efficient synthesis of (S)-4,4-dichloro-3-methylbutanoic acid 8 using organocatalysis and its use in the first total synthesis of dysideaproline E 5 and the diastereomer 7
Results and discussion
Retrosynthetic analysis of dysideaproline E 5 gives three fragments (8–10) which could be coupled via standard peptide chemistry. A similar strategy was proposed for the synthesis of diastereomer 7 assembled on an N-methyl L-leucine core required for comparison with the natural product 5 (Scheme 1).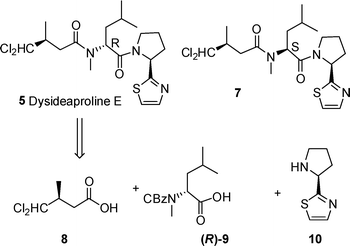 | ||
| Scheme 1 Retrosynthesis of dysideaproline E. | ||
The first goal was to develop an efficient synthetic approach to (S)-dichloro acid 8. Previous syntheses of natural products containing a dichloromethyl group have introduced the halogens either by direct conversion of an aldehyde to a dichloridee.g. in the synthesis of (2S,4S)-5,5-dichloroleucine,6dysithiazolamide,7 sintokamide C8 and nakiteropiosin9 or via enolate chemistry to prepare an α,α-dichloro acid followed by decarboxylation as in the synthesis of dysamide B.10
We proposed that an α,α-dichlorination/decarboxylation strategy could be adopted for the synthesis of the target dichloro acid 8 using a conjugate addition to chiral crotonate 11 to create the required stereocentre (Scheme 2). Thus 11 was treated with allylmagnesium bromide, CuBr·SMe2, LiCl and TMSCl under the conditions reported by Lipshutz and co-workers11 to give the known12alkene 12 in 94% yield. Oxidative cleavage of alkene 12 to aldehyde 13 followed by α,α-dichlorination using N-chlorosuccinimide and tert-butylamine13 gave, after hydrolysis, dichloroaldehyde 14 in good yield. Oxidation of 14 gave acid 15 required for the key decarboxylation step in the presence of a hydride source which would not reduce the dichloride moiety. Treatment of 15 with Pb(OAc)4 and 1,4-cyclohexadiene generated the required dichloride 16 and finally cleavage of the auxiliary gave the target dichloro acid 8 in 7 steps and 18% overall yield from crotonyl sultam 11.
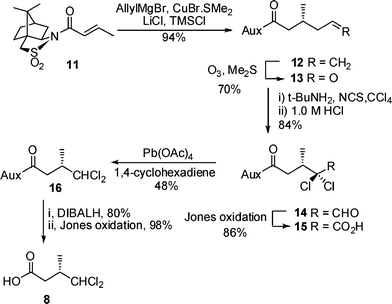 | ||
| Scheme 2 Preparation of (S)-4,4-dichloro-3-methylbutanoic acid 8. | ||
Whilst the above method gave the target 8, a more concise approach was required to prepare large quantities of the dichloro acid for use in total synthesis. Hence a new strategy was proposed involving the asymmetric reduction of an unsaturated carbonyl compound already containing the requisite dichloromethyl group (Scheme 3). However such a reduction would be challenging as it would be necessary to effect a selective asymmetric conjugate reduction without either competing SN2′ attack (leading to formation of a vinyl chloride) or concomitant reduction of the dichloride.
 | ||
| Scheme 3 Proposed new strategy to dichloro acid 8. | ||
Inspired by biological processes which use enzyme mediated reductions with dihydropyridine cofactors such as NADH to perform highly stereoselective reductions, several research groups have successfully replaced the enzymes and co-factors by small molecule organocatalysts and dihydropyridine analogues. For example List and MacMillan have shown that Hantzsch esters can be used in the presence of chiral amine catalysts to reduce β,β-disubstituted-α,β-unsaturated aldehydes in good yields and high enantioselectivities.14 The method is versatile and a series of unsaturated aldehydes with aromatic, heteroaromatic and aliphatic groups at the β-position have been reduced successfully.14,15 Hence we questioned whether the enantio- and chemoselective reduction of a β-dichloromethyl-β-methyl-α,β-unsaturated aldehyde (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, Scheme 3) could be achieved using the biomimetic reaction with an inexpensive hydride source and an air stable chiral catalyst.
H, Scheme 3) could be achieved using the biomimetic reaction with an inexpensive hydride source and an air stable chiral catalyst.
The required substrate 18 was prepared in 2 steps and 62% overall yield using a Horner–Wadsworth–Emmons chain extension of commercially available α,α-dichloroacetone with triethyl phosphonoacetate followed by DIBALH reduction of the resultant ester 17 (Scheme 4). From the 1H-NMR spectrum of 18 it was apparent that a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of isomers had been formed, with the minor isomer showing an nÖe between the methyl group and olefinic proton in accord with the Z-geometry.
:1 mixture of isomers had been formed, with the minor isomer showing an nÖe between the methyl group and olefinic proton in accord with the Z-geometry.
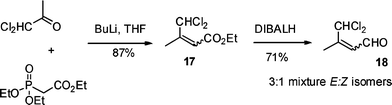 | ||
| Scheme 4 Synthesis of unsaturated aldehyde 18. | ||
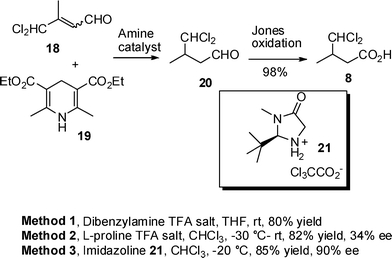
The chemoselectivity of the key reduction step was investigated by treating unsaturated aldehyde 18 with dihydropyridine 19 in the presence of dibenzylamine TFA salt as the catalyst in THF at room temperature (Method 1, Scheme 5). Dichloroaldehyde 20 was isolated as the sole product in a pleasing 80% yield confirming that the required conjugate reduction had occurred selectively. Aldehyde 20 was readily oxidized to acid 8 using Jones reagent. Having established a short and efficient approach to dichloro acid 8, the reduction of 18 was repeated using dihydropyridine 19 and with L-proline TFA salt as a chiral catalyst (Method 2, Scheme 5) and the resultant aldehyde oxidized to acid 8. The enantiopurity of acid 8 was determined by coupling to a thiazolidinethione chiral auxiliary (see ESI†) revealing a 34% ee. Hence a more effective chiral catalyst was required. Imidazolines have been used to good effect in the organotransfer hydrogenation of enals.15,16 In this case reduction of unsaturated aldehyde 18 using imidazolidinone 21 as the catalyst gave significantly better enantioselectivity (90% ee) than with L-proline TFA salt (Method 3, Scheme 5). The absolute configuration was confirmed by comparison with material prepared using the Oppolzer camphorsultam (Scheme 2). Thus a short and efficient synthetic route has been developed giving (S)-dichloro acid 8 in 4 steps and 51% overall yield from commercially available starting materials.
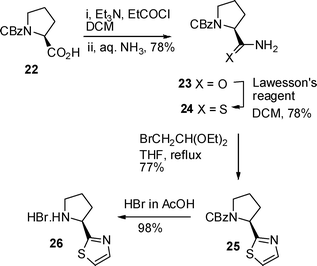
The second component common to both dysideaproline E 5 and its diastereomer 7 was the salt of thiazole 10 which was prepared via a modified Hantzsch method (Scheme 6). First CBz L-proline 22 was converted to amide 23via a mixed anhydride and reaction with aqueous ammonia,17 then treatment with Lawesson's reagent in CH2Cl2 at room temperature18 in dichloromethane gave the required thioamide 24 in 61% overall yield from acid 22. Reaction of thioamide 24 with α-bromoacetaldehyde diethyl acetal in THF at reflux gave thiazole 25 and finally removal of the CBz protecting group with hydrobromic acid yielded the required proline derivative 26.
 | ||
| Scheme 6 Preparation of L-proline thiazoline 26. | ||
To confirm that the stereochemical integrity at C-2 had been maintained throughout the synthetic route the product 26 was compared with racemic material by chiral HPLC. The final compounds needed for the total synthesis of dysideaproline E 5 and diastereomer 7 were N-CBz-N-methyl D- and L- leucines (R-9) and (S-9). We have previously prepared S-9 from L-leucine in two steps and 74% yield viaCBz protection followed by N-methylation with NaH/MeI in MeCN10 and the same approach was used herein to prepare both enantiomers.
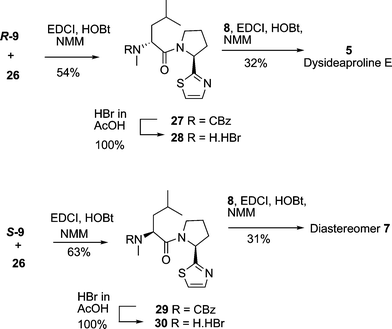
With all the necessary building blocks in hand for the total synthesis of dysideaproline E 5 and the diastereomer 7, the first coupling to be investigated was N-CBz N-methyl D-leucineR-9 with salt 26. Several standard coupling conditions were explored and EDCI, HOBt, NMM in DCM–DMF gave the best yield (54%) of dipeptide 27 (Scheme 7).
 | ||
| Scheme 7 Completing the total synthesis of dysideaproline E 5 and the diastereomer 7. | ||
Reaction of the L-leucine derivativeS-9 with 26 under the same conditions gave 29 in 63% yield. Deprotection of dipeptides 27 and 29 using hydrobromic acid proceeded cleanly to give the salts 28 and 30 in quantitative yield which were coupled with (S)-dichloro acid 8 giving the target compounds 5 and 7. There was an excellent correlation of the 1H- and 13C- NMR data for synthetic 5 with that of the natural product and the optical rotation, [α]D +43.8 (c 0.02, MeOH) was in accord with the literature value3 of [α]D +45.5 (c 0.02, MeOH) for dysideaproline E. In contrast diastereomer 7 assembled on the N-methyl-L-leucine core had an optical rotation [α]D −42.6 (c 1.0, MeOH).
Conclusions
In conclusion two different strategies for the synthesis of (S)-4,4-dichloro-3-methylbutanoic acid 8 have been explored. An operationally simple and efficient approach used an organocatalytic transfer hydrogenation to 4,4-dichloro-3-methylbut-2-enal 18 in the key step giving the target 8 in 51% overall yield from commercially available starting materials. Dichloro acid 8 was used as an intermediate in the first total synthesis of dysideaproline E 5 and its diastereomer 7 confirming the structure of the natural product. This organocatalytic transfer methodology will be of value in the total synthesis of further polychlorinated natural products.Experimental
General details
All commercially available compounds were used without further purification except where stated. All moisture or air sensitive reactions were carried out in oven-dried glassware under a positive pressure of nitrogen using standard syringe/septa techniques. Anhydrous solvents were obtained by passing through a modified Grubbs system of alumina columns, manufactured by Anhydrous Engineering. When stated, DMF was dried sequentially over three portions of 10% w/v 3 Å MS beads for 24 h each, and then stored under nitrogen. Petroleum ether is of the 40–60 °C boiling point range. Routine monitoring of reactions was performed using precoated Merck–Kieselgel 60 F254 aluminium backed TLC plates. The spots were visualised by UV254 light, or potassium permanganate. Flash column chromatography was performed using silica gel (obtained from Fluorochem Ltd.) as the adsorbent. Melting points were determined on an electrothermal apparatus and are uncorrected. Optical rotations were recorded using with the sodium D line (λ = 589 nm) on a Bellingham and Stanley ADP220 polarimeter and the [α]D values are quoted in units 10−1 deg cm2 g−1. Infrared spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer in the solid or liquid state. 1H and 13C NMR spectra were recorded at using either a Jeol Delta/GX 400 MHz or a Jeol Eclipse 400 MHz spectrometer. The chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) are in Hertz (Hz). Tetramethylsilane was used as the internal reference for proton and carbon chemical shifts. DEPT 135, COSY, HBQC and HMBC NMR spectra we were routinely used to definitively assign the signals of 1H and 13C NMR spectra. Electron impact (EI) and chemical ionisation (CI) mass spectra were recorded on a VG Analytical Autospec mass spectrometer. Electrospray (ESI) mass spectra were recorded on a Micromass LCT mass spectrometer or a VG Quattro mass spectrometer. Methane was the ionisation gas used for chemical ionisation.(1S,2R)-N-[(3′R)-Methylhex-5′-enyl]-bornane-10,2-sultam 12
CuBr·Me2S (1.5 eq, 106 mmol, 21.79 g) and LiCl (1.5 eq, 106 mmol, 4.49 g) in THF (100 ml) were pre-stirred for 0.5 h before adding to a solution of allyl magnesium bromide (1.4 eq, 98.84 mmol, 98.84 ml) in THF (300 ml) at −78 °C under N2. TMSCl (1.5 eq, 106 mmol, 13.45 ml) was added to the reaction mixture followed by the immediate addition of the crotonyl auxiliary 11 (20 g, 70.6 mmol) in THF (140 ml). The reaction mixture was stirred for 4 h before quenching with NH4Cl/NH4OH (pH 8, 120 ml) and warming to room temperature overnight. Water (50 ml) and Et2O (50 ml) were added and the layers separated. The aqueous phase was extracted with Et2O (3 × 150 ml) and the combined organic layer was washed with water (2 × 70 ml) and brine (70 ml). The organic layer was dried over anhydrous magnesium sulfate, filtered and the solvent removed in vacuo. Purification by column chromatography (0–5% EtOAc/petrol) gave alkene 12 as a white solid and single diastereomer (21.6 g, 94%); m.p. 74–76 °C; [α]D −75.3 (c 3.4, CHCl3); lit.12b [α]D −88 (c 3.6, CHCl3), δH (400 MHz) 0.95 (3H, d, J 6, CH3), 0.96 and 1.18 (each 3H, s, 8-H3 and 9-H3), 1.2–2.2 (10H, m, 3-H2, 4-H, 5-H2, 6-H2, 3′-H and 4′-H2), 2.49 (1H, dd, J 16, 8, 2′-HH), 2.76 (1H, dd, J 16, 6, 2′-HH), 3.41 and 3.49 (each 1H, d, J 14, 10-H2). 3.87 (1H, t, J 6, 2-H), 5.0 (2H, m, 6′-H2), 5.75 (1H, ddt, J 18, 10, 7, 5′-H); δC (100 MHz) 19.6 (CH3), 19.9 and 20. 9 (C-8 and C-9), 26.5 (C-5), 29.8 (C-3′), 32.9 (C-6), 38.6 (C-2′), 40.9 (C-3), 42.4 (C-4′), 44.8 (C-4), 47.8 (C-1), 48.4 (C-7), 53.1 (C-10), 65.3 (C-2), 116.6 (C-6′), 136.5 (C-5′), 171.5 (CO); νmax/cm−1 3019, 2964, 1720, 1693, 1330 and 1166; m/z (CI) 326 (MH+, 100%), 284 (8), 218 (12), 135 (100); found C 62.4, H 8.72, N 3.45, S 9.91, C17H27NO3S requires C 62.74, H 8.36, N 4.3, S 9.85.(1S,2R)-N-[(3′R)-Methylpent-5′-al]-bornane-10,2-sultam 13
Ozone was bubbled through a solution of alkene 12 (21.6 g, 66.36 mmol) in DCM (200 ml) at −78 °C until a permanent blue colour persisted. DMS (10 eq, 663.7 mmol, 50 ml) was added and the reaction mixture warmed to room temperature before heating under reflux overnight. The reaction mixture was allowed to cool before adding water (50 ml). The layers were separated and the aqueous layer extracted with DCM (3 × 60 ml). The combined organic layer was washed with water (35 ml), dried over anhydrous magnesium sulfate, filtered and the solvent removed in vacuo. Purification by column chromatography (20% EtOAc/petrol) gave aldehyde 13 as a white solid (15.03 g, 70%); m.p. 60–72 °C; [α]D −83.8 (c 2.1, CHCl3); δH (400 MHz) 0.96 and 1.15 (each 3H, s, 8-H3 and 9-H3), 1.05 (3H, d, J 6, CH3), 1.8–2.2 (7H, m, 3-H2, 4-H, 5-H2 and 6-H2), 2.34–2.4 (1H, m, 3-H′), 2.5 (1H, ddd, J 16, 7, 2, 4′-HH), 2.72 (3H, m 4′-HH and 2′-H2), 3.41 and 3.49 (each 1H, d, J 14, 10-H2), 3.87 (1H, t, J 6, 2-H), 9.7 (1H, t, J 2, 5′-H); δC (100 MHz) 14.2 (CH3), 19.9 and 20.8 (C-8 and C-9), 25 (C-3′), 26.5 (C-5), 32.9 (C-6), 38.6 (C-3), 41.9 (C-2′), 44.7 (C-4), 47.8 (C-7), 48.5 (C-1), 50.3 (C-4′), 53 (C-10), 65.3 (C-2), 170.5 and 201.5 (CO); νmax/cm−1 2962, 1705, 1326 and 1166; m/z (CI) 328 (MH+, 53%), 257 (13), 135 (46) and 113 (100); Found C 58.38, H 7.57, N 3.93, S 9.7, C16H25NO4S requires C 58.69, H 7.7, N 4.28, S 9.79.(1S,2R)-N-[(3′S)-Methylpent-4′4′-dichloro-5′-al]-bornane-10,2-sultam 14
tert-Butylamine (1eq, 29.95 mmol, 3.15 g) was added to a solution of aldehyde 13 (9.81 g, 29.95 mmol) in CCl4 (100 ml) in the presence of molecular sieves (4 Å). The reaction mixture was stirred for 1.5 h before adding additional CCl4 (20 ml) and magnesium sulfate. The reaction mixture was filtered and NCS (2.2 eq, 65.89 mmol, 8.80 g) was added in one portion and the reaction mixture stirred overnight. The succinimide was removed by filtration and the filtrate concentrated in vacuo. The product was hydrolysed by stirring in HCl (0.1 M, 260 ml) for 48 h. After this time, Et2O (250 ml) was added and the layers separated. The aqueous layer was extracted with Et2O (3 × 150 ml), the combined organic layers were washed with water (70 ml), dried over anhydrous magnesium sulfate and the solvent removed in vacuo to give dichlorinated aldehyde 14 as a white solid (9.89 g, 84%); m.p. 98–101 °C; [α]D −72 (c 2, CHCl3); δH (400 MHz) 0.99 and 1.1 (each 3H, s, 8-H3 and 9-H3), 1.13 (3H, d, J 7, CH3), 1.2–2.1 (7H, m, 3-H2, 4-H, 5-H2 and 6-H2), 2.8 (1H, dd, J 17, 8, 2′-HH), 3.13 (1H, dd, J 17, 4, 2′-HH), 3.20 (1H, m, 3′-H), 3.45 and 3.53 (each 1H, d, J 14, 10-H2), 3.87 (1H, dd, J 8, 5, 2-H), 9.15 (1H, s, 5′-H); δC (100 MHz) 16.0 (CH3), 19.9 and 20.8 (C-8 and C-9), 26.5 (C-5), 32.9 (C-6), 38.3 (C-3), 38.4 (C-2′), 38.8 (C-3′), 44.7 (C-4), 47.9 (C-7), 48.6 (C-1), 53 (C-10), 65.3 (C-2), 93.5 (C-4′), 169 and 184 (CO); νmax/cm−1 2969, 1740, 1694, 1333 and 1135; m/z (CI) 396 (MH+, 92%), 362 (52), 216 (30), 135 (100); C16H24Cl2NO4 requires 396.0803, found 396.0800.(1S,2R)-N-[(3′S)-4′,4′-Dichloro-3′-methylpentan-5′-oic acid]-bornane-10,2-sultam 15
Dichlorinated aldehyde 14 (7.79 g, 19.66 mmol) was dissolved in acetone (130 ml) and Jones reagent (20 ml) was added. The reaction was monitored by TLC and after 3 h had reached completion. The reaction mixture was quenched with MeOH (25 ml) and the solvent removed in vacuo. Water (40 ml) was added and the aqueous layer extracted with EtOAc (3 × 70 ml). The combined organic layer was washed with water (2 × 30 ml), dried over anhydrous magnesium sulfate and the solvent removed in vacuo. Purification by column chromatography (20–50% EtOAc/petrol) gave acid 15 as a white solid (7.03 g, 86%); m.p. 174–176 °C; [α]D −80.4 (c 2.1, CHCl3), δH (400 MHz) 0.97 and 1.15 (each 3H, s, 8-H3 and 9-H3), 1.2 (3H, d, J 7, CH3), 1.3–2.1 (7H, m, 3-H2,4-H, 5-H2, 6-H2), 2.9 (1H, dd, J 17, 9, 2′-HH), 3.13 (1H, dd, J 17, 2, 2′-HH), 3.3 (1H, m, J 3, 3′-H), 3.44 and 3.54 (each 1H, d, J 14, 10-H2), 3.9 (1H, dd, J 8, 5, 2-H), 9.4 (1H, br s, CO2H); δC (100 MHz) 16.2 (CH3), 19.3 and 20.9 (C-8 and C-9), 26.5 and 26.7 (C-3 and C-5), 32.9 (C-6), 38.4 (C-2′), 41.8 (C-3′), 44.7 (C-4), 47.9 (C-7), 48.6 (C-1), 53.1 (C-10), 65.4 (C-2), 88.8 (C-4′), 167.5 and 169.8 (CO); νmax/cm−1 2971, 1739, 1670, 1371 and 1136; m/z (CI) 412 (MH+, 10%), 376 (6), 339 (8), 295 (46), 135 (100), 216 (40); Found C 46.9, H 5.69, N 3.41, S 7.52, Cl 17.01, C16H23Cl2NO5S requires C 46.61, H 5.62, N 3.4, S 7.78, Cl 17.2.(1S,2R)-N-[(3′S)-4′,4′-Dichloro-3′methylbutane]-bornane-10,2-sultam 16
Dichlorinated acid 15 (0.54 g, 1.3 mmol) was dissolved in MeCN (15 ml) under N2 and stirred for 5 mins. Pb(OAc)4 (1eq, 1.3 mmol, 0.581 g) and 1,4-cyclohexadiene (1.2 eq, 1.6 mol, 0.15 ml) were then added and the reaction mixture heated at reflux for 2 h. After this time the reaction mixture was cooled before adding further Pb(OAc)4 (1 eq) and 1,4-cyclohexadiene (1.2 eq) and heating at reflux for an additional 1 h. The reaction mixture was diluted with Et2O (20 ml) and washed with perchloric acid (7%, 2 × 15 ml). The aqueous layer was extracted with Et2O (2 × 15 ml) and the combined organic layer was washed with NaHCO3 (2 × 20 ml). The aqueous layer was extracted with Et2O (1 × 20 ml) and the combined organic layers were washed with water (10 ml). The combined organic layer was dried over anhydrous magnesium sulfate, filtered and the solvent removed in vacuo. Purification by column chromatography (5–10% EtOAc/petrol) gave dichloride 16 as a white solid (0.32 g, 48%); m.p. 132–135 °C; [α]D −82.9 (c 2.2, CHCl3); δH (400 MHz) 0.97 and 1.15 (each 3H, s, 8 and 9-CH3), 1.2 (3H, d, J 6, CH3), 1.3–2.1 (7H, m, 3-H2, 4-H, 5-H2 and 6-H2), 2.79–2.85 (2H, m, 3-H′ and 2′-HH), 3.0 (1H, dd, J 19, 8, 2′-HH), 3.44 and 3.51 (each 1H, d, J 14, 10-H2), 3.87 (1H, dd, J 8, 5, 2-H), 5.97 (1H, d, J 3, 4′-H); δC (100 MHz) 15.1 (CH3), 19.9 and 20.8 (C-8 and C-9), 26.5, 26.7 and 32.9 (C-3, C-5 and C-6), 38.6 (C-2′), 40.3 (C-3′), 44.7 (C-2), 47.9 (C-4), 48.6 (C-7), 53 (C-10), 65.3 (C-1), 76.7 (C-4′), 169.8 (CO); νmax/cm−1 2963, 1691, 1328 and 1133; m/z (CI) 368 (MH+, 94%), 332 (80), 151 (42) and 135 (100); C15H24Cl2NO3S requires 368.0849, found 368.0853.Ethyl 4,4-dichloro-3-methylbut-2-enoate 17
Triethyl phosphonoacetate (0.78 ml, 3.9 mmol) in THF (10 ml) was cooled to 0 °C under nitrogen and n-butyllithium in hexane (2.5 M, 1.58 ml, 3.9 mmol) was added dropwise. The solution was stirred for 20 min and dichloroacetone (0.5 g, 3.9 mmol) in THF (10 ml) was added dropwise. The solution was stirred at room temperature and monitored by TLC. After 2 h it was quenched with water (15 ml). The mixture was extracted with EtOAc (3 × 30 ml), dried over MgSO4, filtered and the solvent evaporated in vacuo to give the crude product which was purified by column chromatography using EtOAc/petroleum ether 1:9 as the eluent to give ester 17 as a 3:1 mixture of E/Z isomer (0.67 g, 87%); vmax/cm−1 2979, 1745, 1320, 750; major isomer δH (400 MHz, CDCl3), 1.29 (3H, t, J 7.0, OCH2CH3), 2.45 (3H, s, CH3), 4.21 (2H, q, J 7.0, OCH2CH3), 6.0 (1H, br. s), 6.18 (1H, s); δC (100 MHz, CDCl3) 14.2 (CH3), 77.1 (C-4), 118.3 (C-2), 151.9 (C-3), 165.4 (CO); minor isomer, δH (400 MHz, CDCl3), 1.29 (3H, t, J 7.0, OCH2CH3), 2.34 (3H, s, CH3), 4.21 (2H, q, J 7.0, OCH2CH3), 5.71 (1H, s), 7.99 (1H, s); δC (100 MHz, CDCl3) 13.2 (CH3), 67.3 (CHCl2), 118.0 (C-2), 151.7 (C-3), 164.7 (CO); m/z (CI) 201 (4%), 199 (25), 197 (MH+ 38), 196 (8), 161 (68), 133 (69), 85 (65), 83 (100); C7H11O2Cl2 requires 197.0129, found 197.0136.
4,4-Dichloro-3-methylbut-2-enal 18
Diisobutylaluminium hydride (3.28 ml, 3.28 mmol, 1.1 eq) was added to a stirring solution of ester 17 (0.588 g, 2.98 mmol) in dry dichloromethane (10 ml) at −78 °C, under nitrogen over a period of 15 min. The reaction mixture was stirred at −78 °C for 0.5 h and was then quenched with Rochelle's salt (5 ml). The mixture was allowed to stir for 3 h at room temperature, the layers separated and the aqueous layer extracted with ethyl acetate (3 × 10 ml). The organic layers were combined, washed with brine (50 ml) and dried over magnesium sulfate. The solvent was removed in vacuo to give the crude product which was purified using column chromatography eluting with 5% ethyl acetate/petrol to give unsaturated aldehyde 18 as a 3:1 mixture of E/Z isomers as yellow oil (0.32 g, 71%); vmax/cm−1 2789, 1738, 1645, 750; major isomer, δH (400 MHz, CDCl3), 2.45 (3H, d, J 1.3, CH3), 6.08 (1H, br d, J 7.3, 2-H), 6.18 (1H, s, 4-H) 10.06 (1H, d, J 7.3, 1-H); δC (100 MHz, CDCl3) 14.2 (CH3), 79.6 (C-4), 128.7 (C-2), 156.3 (C-3), 193.3 (CO); minor isomer, δH (400 MHz, CDCl3), 2.34 (3H, d, J 1.3, CH3), 5.94 (1H, br. d, J 5.3, 2-H), 7.46 (1H, s, 4-H), 9.90 (1H, d, J 5.3, 1-H); δC (100 MHz, CDCl3) 13.2 (CH3), 79.0 (CHCl2), 128.5 (C-2), 155.5 (C-3), 189.4 (CO); m/z (CI) 157 (8%),155 (22), 153 (MH+ 58), 117 (85), 113 (52), 97 (79), 89 (100); C5H7O35Cl2 requires 152.9870, found 152.9874.
(S)-4,4-Dichloro-3-methylbutanal 20
A solution of unsaturated aldehyde 18 (0.814 g, 5.32 mmol) dissolved in chloroform (5 ml) was cooled to −20 °C in a dry ice/acetone bath. To this solution was added the trichloroacetic acid salt of (R)-2-tert-butyl-3-methylimidazolidin-4-one 21 (0.287 g, 1.06 mmol) and Hantzsch ester 19 (1.98 g, 6.33 mmol). The resulting yellow suspension was stirred at −20 °C for 4 h and allowed to warm room temperature overnight. The reaction mixture was poured into HCl solution (10%, 30 ml) and diluted with ether (30 ml). The organic layer was washed 4 times with HCl solution (10%, 30 ml) and once with a saturated aqueous solution of NaHCO3 (30 ml). The resulting organic solution was dried with magnesium sulfate and concentrated in vacuo. The residue was purified by flash chromatography using 5% ether in pentane to give aldehyde 20 as a yellow oil (0.694 g, 85%); [α]23D −6.0 (c 1, CHCl3). vmax/cm−1 2789, 1730, 750; δH (270 MHz, CDCl3), 1.25 (3H, d, J 6.8, CH3), 2.45 (1H, dd, J 10.5, 7.3, 2-HH), 2.84–2.95 (2H, m, 2-HH, 3-H), 5.89 (1H, d, J 3.3, 4-H), 9.81 (1H, br. s, 1-H), δC (67.5 MHz, CDCl3), 15.8 (CH3), 38.4 (C-3), 46.4 (C-2), 76.6 (CHCl2), 199.8 (C-1), m/z (CI) 157 (5%), 155 (MH+ 10), 117 (55), 89 (55), 83 (100); C5H9OCl2 requires 155.0025, found 155.00304,4-Dichloro-3-methylbutanoic acid 8
Jones reagent was prepared by dissolving chromium trioxide (25 g, 250 mmol) in a solution of concentrated sulfuric acid (20 ml) and distilled water (70 ml). Aldehyde 20 (0.077 g, 0.49 mmol) was dissolved in acetone (10 ml) and Jones reagent was added dropwise until an orange colour persisted. The reaction was stirred for 15 min, after which time methanol (5 ml) was added dropwise to quench the reaction followed by water (5 ml). The solvent was removed in vacuo and the green-black residue diluted with water (5 ml) and extracted with ethyl acetate (3 × 10 ml). The combined organic extract was dried over anhydrous MgSO4, filtered and the solvent removed in vacuo. Purification by column chromatography (20% EtOAc/petrol + 1% AcOH) gave dichlorinated acid 8 as a clear liquid (0.083 g, 98%); [α]23D −8.2 (c 1, CHCl3), [α]23D −7.8 (c 1, Et2O) vmax/cm−1 3400–3200, 2979, 1705, 750; δH (270 MHz, CDCl3), 1.25 (3H, d, J 6.9, CH3), 2.45 (1H, dd, J 10.5, 7.3,2-HH), 2.61–2.80 (2H, m, 2-HH, 3-H), 5.89 (1H, d, J 3.3, 4-H), 10.5 (1H, br s, OH), δC (100 MHz, CDCl3), 15.4 (CH3), 36.7 (C-3), 40.5 (C-2), 76.6 (CHCl2), 178.1 (C-1), m/z (CI) 175 (2%) 173 (10), 171 (MH+ 15),135 (35), 99 (100), 83 (65); C5H9O235Cl2 requires 170.9972, found 170.9980N-Methyl-N-(benzyloxycarbonyl)-D-leucine (R)-9
Cbz-D-Leucine (5 g, 18 mmol) was dissolved in acetonitrile (60 ml) and cooled to 0 °C. Sodium hydride (2.17 g, 56.5 mmol) was added followed by dropwise addition of methyl iodide (8 ml, 128 mmol) to form a thick liquid. The reaction mixture was stirred overnight. EtOAc (70 ml) and water (25 ml) were added and the solvent evaporated off. The crude mixture was dissolved in ether (30 ml) and water (30 ml), the layers were separated and the aqueous layer was washed with ether (2 × 20 ml). The organic layers were combined and washed with saturated NaHCO3 (2 × 15 ml). All the aqueous layers were combined and acidified to pH 2 using HCl (1 M), extracted with EtOAc (3 × 35 ml) and all organic layers were combined and washed with water (15 ml), dried over magnesium sulfate and the solvent evaporated to give a dark orange viscous oil. This was purified by column chromatography (loaded on in DCM) using eluent 20% EtOAc:petrol to give (R)-9 as a white solid (4.7 g, 90% yield); mp 64–69 °C (no lit. mp reported); [α]23D + 25.4 (c 1, EtOH) [lit.10 +23 (c 1, EtOH)]; δH (400 MHz), major rotamer) 0.95 and 0.98 (each 3H, d, J 6, 2 × CH3), 1.58 (1H, m, 4-H), 1.63–1.80 (2H, m, 3-H2), 2.89 (3H, s, N-CH3), 4.93 (1H, t, J 7.9 (minor rotamer, dd, J 10,5 and 4.5), 2-H), 5.1–5.2 (2H, m, OCH2), 7.3–7.4 (5H, m, PhH); δC (100 MHz both rotamers) 21.0 and 21.3 (CH3), 23.1 (CH3), 24.7 and 24.9 (C-4), 30.4 and 30.7 (NCH3), 37.3 and 37.7 (C-3), 56.6 and 56.8 (C-2), 67.6 (OCH2) 127.7–128.4 (PhH), 136.5 (ipso), 157.2 and 177.3 (CO).N-(Benzyloxycarbonyl)-L-proline thiazole 25
2-((S)-2-Pyrrolidinyl)-1,3-thiazole hydrobromic salt 2619
Thiazole 25 (222 mg, 0.77 mmol) was dissolved in HBr (3 ml, 33% in acetic acid) and stirred for 4 h at room temperature. The reaction mixture was concentrated in vacuo and dried in the vacuum oven at 60 °C overnight to give salt 26 as an orange solid (179 mg, 98%); [α]23D +28.5 (c 1, H2O); vmax/cm−1 2860, 2440, 1564, 1368 and 1286 δH (400 MHz, D2O) 2.21–2.48 (3H, m, 3HH and 4-H2), 2.65–2.77 (1H, m, 3HH), 3.54–3.63 (2H, m, 5-H2), 5.23 (1H, t, J 7.8, 2-H), 7.77 (1H, d, J 3.4, 7-H), 7.93 (1H, d, J 3.4, 6-H); δC (100 MHz) 23.5 (C-4), 30.9 (C-3), 45.9 (C-5), 59 (C-2), 122.3 (C-7), 142.8 (C-6), and 164.4 (C-1); m/z (ESI) 289 (MH+); C15H17O2NS requires 289.0999 found 289.1011.N-Methyl-[(R)-3-methyl-1-((S)-thiazol-2-yl-pyrrolidine-1-carbonyl)-butyl]-carbamic acid benzyl ester 27
L-Proline thiazole hydrobromic salt 26 (170 mg, 0.72 mmol) and N-Cbz-N-Me-D-leucine (R)-9 (180 mg, 0.72 mmol) formed a suspension with DCM–DMF (5:1 ratio, 11 ml). The reaction mixture was cooled to 0 °C and EDCI (2 eq, 277 mg, 1.45 mmol), HOBt (1.1 eq, 108 mg, 0.79 mmol) and NMM (1.5 eq, 0.12 ml, 1.08 mmol) were added and the reaction mixture was stirred at 0 °C for 1 h and then at room temperature overnight. The reaction was quenched with HCl (1 M, 5 ml) and diluted with EtOAc (15 ml). The mixture was separated and the organic layer was then washed with NaHCO3 (10 ml), brine (10 ml), water (10 ml), dried over magnesium sulfate and the solvent evaporated in vacuo to give the crude product which was purified with flash chromatography eluting with 20% acetone/petrol to give 27 as viscous white oil (156 mg, 54%); [α]23D +18.8 (c 1, MeOH); vmax/cm−1 2955, 2875, 1697, 1654; δH (400 MHz, major rotamer) 0.95 and 0. 97 (6H, 2 × d, J 6.6, 2 × CH3), 1.3 and 1.73 (2H, m, 3-H2), 1.5 (1H, m, 2-H), 1.97 (2H, m, 10-H2), 2.05–2.39 (2H, m, 9-H2), 2.83 (3H, s, N-CH3), 3.45–3.78 (2H, m, 11-H2), 5.09 (1H, m, 4-H), 5.22 (2H, m, OCH2), 5.5 (1H, dd, J 7.9, 2.8, 8-H), 7.17 (1H, d, J 3.3, 14-H), 7.67 (1H, d, J 3.3, 13-H); δC (100 MHz major rotamer) 22.3 (C-1), 23.0 (C-7), 24.4 (C-10), 24.5 (C-2), 29.3 (C-6), 31.5 (C-9), 37.3 (C-3), 46.7 (C-11), 54.6(C-4), 58.6 (C-8), 67.3 (OCH2), 118.7 (C-14), 127.6-128.8 (PhH), 136.7 (ipso-C), 142.2 (C-13), 156.6 (CO2CH2), 170 (CO), 172.2 (C-12), m/z (ESI) 416 (MH+); C22H30O3N3S requires 416.2008 found 416.2005.
N-Methyl-[(S)-3-methyl-1-((S)-thiazol-2-yl-pyrrolidine-1-carbonyl)-butyl]-carbamic acid benzyl ester 29
The above method was repeated with N-Cbz-N-Me-L-leucine(S)-9 to give 29 as a viscous yellow oil (182 mg, 63%); [α]23D −99.1 (c 1, MeOH); vmax/cm−1 2955, 2875, 1697, 1654; δH (400 MHz, major rotamer) 0.95 and 0. 97 (6H, 2 × d, J 6.6, 2 × CH3), 1.57 (1H, m, 2-H), and 1.56 and 1.72 (2H, m, 3-H2), 1.5–1.64 and 2.0–2.16 (2H, m, 10-H2), 1.96–2.08 and 2.2–2.35 (2H, m, 9-H2), 2.05–2.39 (2H, m, 9-H2), 2.97 (3H, s, N-CH3), 3.78 (2H, m, 11-H2), 5.12 (1H, dd, J 9.7, 5.2, 4-H), 5.17 (2H, m, OCH2), 5.5 (1H, dd, J 7.9, 2.8, 8-H), 7.17 (1H, d, J 3.3, 14-H), 7.67 (1H, d, J 3.3, 13-H); δC (100 MHz major rotamer) 22.3 (C-1), 23.0 (C-7), 24.4 (C-10), 24.5 (C-2), 29.3 (C-6), 31.5 (C-9), 37.3 (C-3), 46.7 (C-11), 54.6(C-4), 58.6 (C-8), 67.3 (OCH2), 118.7 (C-14), 127.6–128.8 (PhH), 136.7 (ipso-C), 142.2 (C-13), 156.6 (CO2CH2), 170 (CO), 172.2 (C-12), m/z (ESI) 416 (MH+); C22H30O3N3S requires 416.2008 found 416.2005.4-(R)-4-N-Methyl-2-methylamino-1-((S)-2-thiazol-2-yl-pyrrolidin-1-yl)-pentan-1-one 28
Amide 27 (167 mg, 0.4 mmol) was dissolved in HBr (2.5 ml, 33%w/w in acetic acid) and stirred at room temperature for 4 h. The acetic acid was evaporated off and the residue dried in the vacuum oven at 60 °C for 3 days to give 28 as an orange sticky solid in quantitative yield (139 mg, 100%); vmax/cm−1 3323, 2955, 1651, 1551, 1385 and 1102; δH (400 MHz, D2O) 1.06 and 1.09 (6H, 2 × d, J 6, 2 × CH3), 1.86 (3H, m, 2-H and 3-H2), 2.24 (3H, m, 9HH and 10-H2), 2.55 (1H, m, 9HH), 2.74 (3H, s, N-CH3), 3.78 (1H, m, 11-HH), 4.16 (1H, m, 11-HH), 4.41 (1H, dd, J 7.6, 5, 4-H), 5.61 (1H, dd, J 8, 2.4, 8-H), 7.74 (1H, d, J 3.5, 14-H), 7.92 (1H, d, J 3.5,13-H); δC (100 MHz) 20.91 (C-1), 22.23 (C-7), 23.54 (C-10), 23.85 (C-2), 32.07 (C-6), 32.29 (C-9), 38.39 (C-3), 47.76 (C-11), 59.11(C-4), 59.65 (C-8), 67.5 120.9 (C-14), 139.6 (C-13), 168.0 (CO), 173.67 (CN), m/z (ESI) 282 (MH+); C14H24ON3S requires 282.1640 found 282.1637.4-(S)-4-N-Methyl-2-methylamino-1-((S)-2-thiazol-2-yl-pyrrolidin-1-yl)-pentan-1-one 30
The above method was repeated with 29 (151 mg, 0.376 mmol) to give 30 as an orange sticky solid (130 mg, 100%); vmax/cm−1 3387, 2959, 1643, 1551, 1332 and 1111; [α]23D −57.9 (c 1, H2O); δH (400 MHz, D2O) 1.03 and 1.07 (6H, 2 × d, J 6, 2 × CH3), 1.83 (3H, m, 2-H and 3-H2), 2.25 (3H, m, 9HH and 10-H2), 2.57 (1H, m, 9HH), 2.80 (3H, s, N-CH3), 3.96 (2H, m, 11-H2), 4.38 (1H, t, J 6.5, 4-H), 5.64 (1H, dd, J 8.5, 3.7, 8-H), 7.70 (1H, d, J 3.5, 14-H), 7.90 (1H, d, J 3.5,13-H); δC (100 MHz) 21.1 (C-1), 21.8 (C-7), 23.9 (C-2), 24.05 (C-10), 32.0 (C-6), 32.1 (C-9), 38.8 (C-3), 48.0 (C-11), 58.8 (C-4), 59.0 (C-8), 67.5 120.0 (C-14), 140.6 (C-13), 168.4 (CO), 173.2 (CN), m/z (ESI) 282 (MH+); C14H24ON3S requires 282.1640 found 282.1637.Dysideaproline E 5
L-Proline thiazole hydrobromic salt 28 (157 mg, 0.45 mmol) and (S)-4,4-dichloro-3-methylbutanoic acid 8 (116 mg, 0.676 mmol, 1.5 eq) formed a suspension with DCM–DMF (5:1 ratio, 11 ml). The reaction mixture was cooled to 0 °C and EDCI (2 eq, 173 mg, 0.9 mmol), HOBt (1.1 eq, 67 mg, 0.49 mmol) and NMM (1.5 eq, 0.1 ml, 0.676 mmol) were added and the reaction mixture was stirred at 0 °C for 1 h and then at room temperature overnight. The reaction was quenched with HCl (1 M, 5 ml) and diluted with EtOAc (15 ml). The mixture was separated and the organic layer was then washed with NaHCO3 (10 ml), brine (10 ml), water (10 ml), dried over MgSO4 and solvent evaporated to give crude 5 which was purified with flash chromatography eluting with 20% acetone/petrol to give dysideaproline E 5 as viscous clear oil (66 mg, 32%); [α]23D +43.8 (c 0.02, MeOH), [lit.3 +45.5 (c 0.02, MeOH)]; vmax/cm−1 2955, 2871, 1695, 1650, 1453 and 1153; δH (400 MHz, major rotamer) 0.87 (3H, d, J 7.0, CH3), 0. 88 (3H, d, J 7.0, CH3), 1.09 (3H, d, J 6.6, CH3), 1.35 (1H, m, 7-H), 1.51 (1H, m, 6HH), 1.51 (1H, m, 6HH), 1.89 (1H, m, 15HH), 1.96 (1H, m, 15HH), 2.06 (1H, m, 14HH), 2.23 (1H, m, 14HH), 2.46 (1H, dd, J 7.6, 16.3, 3HH), 2.57 (1H, dd, J 7.6, 16.3, 3HH), 2.69 (1H, m, 2H), 2.81 (3H, s, N-CH3), 3.40 (1H, m, 16HH), 3.55 (1H, m, 16HH), 5.30 (1H, dd, J 3.2, 8.2, 13-H), 5.37 (1H, dd, J 6.6, 8.3, 5-H), 6.41 (1H, d, J 3.2, 11-H), 7.57 (1H, d, J 3.3, 19-H), 7.70 (1H, d, J 3.3, 18-H); δC (100 MHz, DMSO) 14.5 (C-1), 22.1 (C-9), 23.0 (C-8), 23.9 (C-15), 24.0 (C-7), 30.2 (C-10), 31.5 (C-14), 35.6 (C-3), 36.9 (C-6), 40.2 (C-2), 46.4 (C-16), 51.7 (C-5), 58.4 (C-13), 79.1 (C-11), 119.5 (C-19), 142.1 (C-18), 169.2 (C-4), 170.2 (C-12) and 172.6 (C-17); m/z 438 (2%),436 (6),434 (10), 362(45), 280 (100), 100 (65); C19H30N3O2S35Cl2 requires 434.1425 found 434.1436.
Diastereomer 7
L-Proline thiazole hydrobromic salt 30 (157 mg, 0.45 mmol) and (S)-4,4-dichloro-3-methylbutanoic acid 8 (116 mg, 0.676 mmol, 1.5 eq) formed a suspension with DCM–DMF (5:1 ratio, 11 ml). The reaction mixture was cooled to 0 °C and EDCI (2 eq, 173 mg, 0.9 mmol), HOBt (1.1 eq, 67 mg, 0.49 mmol) and NMM (1.5 eq, 0.1 ml, 0.676 mmol) were added and the reaction mixture was stirred at 0 °C for 1 h and then at room temperature overnight. The reaction was quenched with HCl (1 M, 5 ml) and diluted with EtOAc (15 ml). The mixture was separated and the organic layer was then washed with NaHCO3 (10 ml), brine (10 ml), water (10 ml), dried over MgSO4 and solvent evaporated to give crude 7 which was purified with flash chromatography eluting with 20% acetone/petrol to give 7 as viscous clear oil (61 mg, 31%); [α]23D −42.6 (c 1, MeOH); 2955, 2871, 1695, 1650, 1453 and 1153; δH (400 MHz, major rotamer), 0.87 (3H, d, J 7.0, CH3), 0. 88 (3H, d, J 7.0, CH3), 1.09 (3H, d, J 6.6, CH3), 1.35 (1H, m, 7-H), 1.51 (1H, m, 6HH), 1.51 (1H, m, 6HH), 1.89 (1H, m, 15HH), 1.96 (1H, m, 15HH), 2.06 (1H, m, 14HH), 2.23 (1H, m, 14HH), 2.46 (1H, dd, J 7.6, 16.3, 3HH), 2.57 (1H, dd, J 7.6, 16.3, 3HH), 2.69 (1H, m, 2H), 2.81 (3H, s, N-CH3), 3.40 (1H, m, 16HH), 3.55 (1H, m, 16HH), 5.30 (1H, dd, J 3.2, 8.2, 13-H), 5.37 (1H, dd, J 6.6, 8.3, 5-H), 6.41 (1H, d, J 3.2, 11-H), 7.57 (1H, d, J 3.3, 19-H), 7.70 (1H, d, J 3.3, 18-H); δC 14.5 (C-1), 22.1 (C-9), 23.0 (C-8), 23.9 (C-15), 24.0 (C-7), 30.2 (C-10), 31.5 (C-14), 35.6 (C-3), 36.9 (C-6), 40.2 (C-2), 46.4 (C-16), 51.7 (C-5), 58.4 (C-13), 79.1 (C-11), 119.5 (C-19), 142.1 (C-18), 169.2 (C-4), 170.2 (C-12) and 172.6 (C-17); m/z 438 (2%),436 (6),434 (10), 362(45), 280 (100), 100 (65); C19H30N3O2S35Cl2 requires 434.1425 found 434.1436
Acknowledgements
We are very grateful to Drs George G. Harrigan, Gilles H. Goetz and Shengtian Yang for supplying copies of the NMR spectra of the natural product dysideaproline E and to the following for funding: BBSRC (ACD), EPSRC (SJO), AstraZeneca (ACJ) and to the Government of Ghana for a scholarship (EOA)Notes and references
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364 CrossRef; G. W. Gribble, J. Chem. Educ., 2004, 81, 1441 CrossRef CAS.
- D. P. Galonić, F. H. Vaillancourt and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 3900 CrossRef CAS; P. M. Flatt, S. J. O'Connell, K. L. McPhail, G. Zeller, C. L. Willis, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2006, 69, 938 CrossRef CAS; C. D. Murphy, Nat. Prod. Rep., 2006, 23, 147 RSC.
- G. G. Harrigan, G. H. Goetz, H. Luesch, S. Yang and J. Likos, J. Nat. Prod., 2001, 64, 1133 CrossRef CAS.
- See for example: J. B. MacMillan and T. Molinski, J. Nat. Prod., 2000, 63, 155 Search PubMed; J.-Y. Su, Y.-L. Zhong, L.-M. Zheng, S. Wei, Q.-W. Wang, T. C. W. Mak and Z.-Y. Zhou, J. Nat. Prod., 1993, 56, 637 CrossRef CAS; B. L. Stapleton, G. M. Cameron and M. J. Garson, Tetrahedron, 2001, 57, 4603 CrossRef CAS; M. D. Unson, C. B. Rose, D. J. Faulkner, L. S. Brinen, J. R. Steiner and J. Clardy, J. Org. Chem., 1993, 58, 6336 CrossRef CAS.
- M. D. Sadar, D. E. Williams, N. R. Mawji, B. O. Patrick, T. Wikanta, E. Chasanah, H. E. Irianto, R. Van Soest and R. J. Andersen, Org. Lett., 2008, 10, 4947 CrossRef CAS.
- A. Ardá, C. Jiménez and J. Rodríguez, Tetrahedron Lett., 2004, 45, 3241 CrossRef CAS; A. C. Durow, C. Butts and C. L. Willis, Synthesis, 2009, 2854.
- A. Ardá, J. Rodríguez, R. Nieto, C. Bassarello, L. Gomez-Paloma, G. Bifulco and C. Jiménez, Tetrahedron, 2005, 61, 10093 CrossRef CAS; A. Ardá, R. G. Soengas, M. I. Nieto, C. Jiménez and J. Rodríguez, Org. Lett., 2008, 10, 2175 CrossRef CAS.
- Y. Jin, Y. Liu, Z. Wang, S. Kwong, Z. Xu and T. Ye, Org. Lett., 2010, 12, 1100 CrossRef CAS.
- S. Gao, Q. Wang and C. Chen, J. Am. Chem. Soc., 2009, 131, 1410 CrossRef CAS; S. Gao, Q. Wang, L. J.-S. Huang, L. Lum and C. Chen, J. Am. Chem. Soc., 2010, 132, 371 CrossRef CAS.
- A. C. Durow, G. C. Long, S. J. O'Connell and C. L. Willis, Org. Lett., 2006, 8, 5401 CrossRef CAS.
- B. H. Lipshutz and C. Hackmann, J. Org. Chem., 1994, 59, 7437 CrossRef CAS.
- (a) S. L. Bouet and L. A. Paquette, Synthesis, 2002, 895 Search PubMed; (b) H. Yang, R. G. Carter and L. N. Zakharov, J. Am. Chem. Soc., 2008, 130, 9238 CrossRef CAS.
- R. Verhé, N. De Kimpe, L. De Buyck and N. Schamp, Synthesis, 1975, 455 CrossRef CAS.
- L. H. P. Meijer and U. K. Pandit, Tetrahedron, 1985, 41, 467 CrossRef; J. W. Yang, M. T. Hechavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660 CrossRef; Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS; J. W. Yang, M. T. Hechavarria Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108 CrossRef CAS; S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS; S. Mayer and B. List, Angew. Chem., Int. Ed., 2006, 45, 4193 CrossRef CAS; J. B. Tuttle, S. G. Oulett and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662 CrossRef CAS.
- T. J. Hoffman, J. Dash, J. H. Rigby, S. Arseniyadis and J. Cossy, Org. Lett., 2009, 11, 2756 CrossRef CAS.
- S. G. Ouellet, A. M. Walji and D. W. C. MacMillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS.
- Y. Seto, K. Torii, K. Bori, K. Inabata, S. Kuwata and H. Watanabe, Bull. Chem. Soc. Jpn., 1974, 47, 151 CrossRef CAS.
- C. J. Moody and M. C. Bagley, J. Chem. Soc., Perkin Trans. 1, 1998, 601 RSC.
- Thiazole 26 has been reported previously but no experimental details or spectroscopic data were recorded: J. Lloyd, H. J. Fimlay, W. Vacarro, A. Kover, R. Bhandaru, L. Yan, K. Atwawl, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li and H. Sun, Bioorg. Med. Chem. Lett., 2010, 20, 1436 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of the enantiopurity of acid 8; NMR spectra of 7, 8, 18, 20 and dysideaproline E 5. See DOI: 10.1039/c0ob00617c |
| This journal is © The Royal Society of Chemistry 2011 |