Total synthesis of (−)-20-epiuleinevia stereocontrolled one-pot asymmetric azaelectrocyclization followed by novel 1,4-addition reaction†
Taku
Sakaguchi
,
Shohei
Kobayashi
and
Shigeo
Katsumura
*
School of Science and Technology, Kwansei Gakuin University, Gakuen 2-1, Sanda, Hyogo 669-1337, Japan. E-mail: katsumura@kwansei.ac.jp; Fax: +81 (79) 565-9077; Tel: +81 (79) 565-8314
First published on 17th November 2010
Abstract
The first asymmetric total synthesis of an indole alkaloid, (−)-20-epiuleine, containing the 2,3,4-trisubstituted piperidine core, was achieved using a stereocontrolled one-pot asymmetric 6π-azaelectrocyclization followed by a stereoselective 1,4-addition reaction of the unsaturated ester with a Grignard reagent resulting from the novel neighboring participation of the hydroxyl group in cis-aminoindanol as a chiral nitrogen source.
Introduction
Uleine (1), 20-epiuleine (2), dasycarpidone (3) and 20-epidasycarpidone (4) are four related members of the Strychnos-type indole alkaloids, which were principally isolated from Aspidosperma sp (Fig. 1.).1 These indole alkaloids lack the two-carbon chain from tryptophan. Although a number of synthetic efforts for the racemic 20-epiuleine (2) and epidasycarpidone (4) along with uleine (1) and dasycarpidone (3) were reported in the literature,2,3 only a few asymmetric syntheses of these indole alkaloids have been reported.4,5 The structural feature of these alkaloids is the 2-aza-bicyclo[3,3,1]nonane skeleton, which has been constructed from the 3-(2-piperidyl)indole derivative 5 by an acid-catalyzed cyclization followed by dehydration (Scheme 1).3 The subject, therefore, is how to efficiently synthesize the optically homogeneous 2-indolyl-3-ethyl-4-carbonyl piperidine compound such as 5, which is classified as a 2,3,4-trisubstituted piperidine skeleton and was obtained in the dl-form by the 1,4-addition of the unsaturated ketone 6 (Scheme 1).3c,3d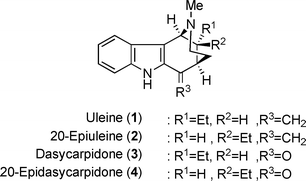 | ||
| Fig. 1 Strychnos-type indole alkaloids. | ||
 | ||
| Scheme 1 Synthetic strategy of 20-epiuleine. | ||
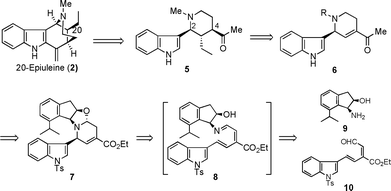
Previously, we reported the formal synthesis of the optically active 20-epiuleine (2) utilizing the step-wise asymmetric 6π-azaelectrocyclization, which was developed as a synthetic strategy for substituted piperidines.5 This asymmetric azaelectrocyclization includes highly controlling the stereochemistry at the second position of the constructed tetrahydropyridine derivative, such as 7, resulting from a kinetically controlled torquoselective 6π-azaelectrocyclization of a conformationally flexible linear azatriene, such as 8, prepared from cis-aminoindanol derivative 9 and dienal 10.5 In that formal synthesis, although we synthesized the optically homogeneous 2-indolyl-4-acyl-1,2,5,6-tetrahydropyridine 6, which was identical to the synthetic intermediate of (dl)-20-epiuleine reported by Husson's group, the transformation to the corresponding chiral 2,3,4-trisubstituted piperidine derivative 5 was not realized. Thus, the real procedure for the construction of the chiral 2,3,4-trisubstituted piperidine framework has not been yet established.
Most recently, in order to establish asymmetric 6π-azaelectrocyclization as a new strategy for alkaloid synthesis, we realized a one-pot protocol, which enabled the facile and highly stereoselective preparation of chiral 2,4-disubstituted- and 2,4,5-trisubstituted-1,2,5,6-tetrahydropyridine derivatives such as 13 under thermodynamic control (Scheme 2).6 Based on the developed protocol, we achieved the stereoselective total synthesis of (−)-dendroprimine,7 which possesses a chiral 2,4,6-trisubstituted piperidine nucleus, and (−)-corynantheidol,8 which has a chiral 2,4,5-trisubstituted piperidine structural unit. In this paper, we describe in detail the stereoselective total synthesis of (−)-20-epiuleine, which consists of a 2,3,4-trisubstituted piperidine structural unit having an indole nucleusvia the one-pot asymmetric azaelectrocyclization followed by the unique 1,4-addition reaction of the unsaturated ester with Grignard reagents resulting from the novel neighboring participation of the hydroxyl group in cis-aminoindanol, which was utilized as a chiral nitrogen source.
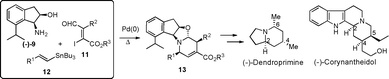 | ||
| Scheme 2 Alkaloid synthesis via 6π-azaelectrocyclization. | ||
Results and Discussion
Usual 1,4-additon reaction of unsaturated methyl ketone with cuprate
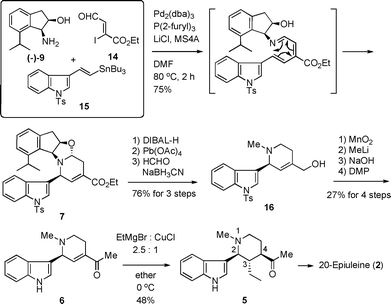
As previously reported, the tetracyclic aminoacetal derivative 7 was stereoselectively synthesized by the one-pot asymmetric 6π-azaelectrocyclization (Scheme 3).6 Thus, the cis-aminoindanol derivative (−)-9, trisubstituted vinyl iodide 14 and MS4A were stirred in DMF at room temperature. After checking the consumption of 14 by TLC, indolyl vinyl stannane 15, Pd2(dba)3, tri(2-furyl)phosphine and LiCl were added, and the resulting mixture was heated at 80 °C for 2 h to produce the desired aminoacetal 7 in 75% yield resulting from the successive Migita-Stille coupling, 1-azatriene formation, 6π-azaelectrocyclization and aminoacetal formation. The stereoselectivity at the second position and the total yield were better than those obtained by the previously developed stepwise procedure (75% yield and >16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stereoselectivity for the one-pot protocol vs. 62% yield and 10:1 stereoselectivity for the stepwise method).5,6 The transformation from 7 to methyl ketone 6 through 16 was accomplished by the sequence of DIBAL-H reduction, lead tetraacetate oxidation in the presence of n-propylamine, reductive N-methylation (HCHO, NaBH3CN in CH3CN), MnO2 oxidation, methylation with MeLi, hydrolysis of the N-tosyl group in the indole ring and then DMP oxidation as previously reported.5 We thus obtained the optically active methyl ketone derivative 6, which was a key synthetic intermediate of (±)-20-epiuleine as reported by Husson's group. We then attempted the 1,4-addition reaction of (−)-6 under the reaction conditions described in the literature.3c After a detailed investigation of the reaction conditions, we obtained the desired 2,3,4-trisubstituted piperidine compound 5 in 48% yield using excess reagents of ethylmagnesium bromide and copper(I) chloride in a ratio of 2.5:1 in ether. When the one to one ratio of ethylmagnesium bromide and copper(I) was used, we did not obtained the desired product and recovered the starting materials. As the solvent of this 1,4-addition reaction, the yield in ether was rather better than in THF. Although we obtained the derived 5, this 1,4-addition reaction was troublesome and sometimes not reproducible. The yield varied from 48% to 13% under the same conditions employed. We then examined another method for this 1,4-addition reaction.
:1 stereoselectivity for the one-pot protocol vs. 62% yield and 10:1 stereoselectivity for the stepwise method).5,6 The transformation from 7 to methyl ketone 6 through 16 was accomplished by the sequence of DIBAL-H reduction, lead tetraacetate oxidation in the presence of n-propylamine, reductive N-methylation (HCHO, NaBH3CN in CH3CN), MnO2 oxidation, methylation with MeLi, hydrolysis of the N-tosyl group in the indole ring and then DMP oxidation as previously reported.5 We thus obtained the optically active methyl ketone derivative 6, which was a key synthetic intermediate of (±)-20-epiuleine as reported by Husson's group. We then attempted the 1,4-addition reaction of (−)-6 under the reaction conditions described in the literature.3c After a detailed investigation of the reaction conditions, we obtained the desired 2,3,4-trisubstituted piperidine compound 5 in 48% yield using excess reagents of ethylmagnesium bromide and copper(I) chloride in a ratio of 2.5:1 in ether. When the one to one ratio of ethylmagnesium bromide and copper(I) was used, we did not obtained the desired product and recovered the starting materials. As the solvent of this 1,4-addition reaction, the yield in ether was rather better than in THF. Although we obtained the derived 5, this 1,4-addition reaction was troublesome and sometimes not reproducible. The yield varied from 48% to 13% under the same conditions employed. We then examined another method for this 1,4-addition reaction.
 | ||
| Scheme 3 Synthesis of 20-epiuleine through unsaturated methyl ketone 6. | ||
Novel 1,4-addition reaction of the unsaturated ester with the Grignard reagent to produce 2,3,4-trisubstituted piperidine derivatives; Improved synthesis of (−)-20-epiuleine
Since it was preferable to construct the 2,3,4-trisubstituted piperidine nucleus of 20-epiuleine at the earlier stage, we attempted the 1,4-addition reaction of the aminoacetal derivative 7, which was easily obtained by the one-pot asymmetric azaelectrocyclization of the three components.6 Surprisingly, the treatment of 7 with an excess amount of the Grignard reagent at 0 °C spontaneously provided the dialkylated ester 17 as a mixture of stereoisomers, and no 1,4-addition product 18 was detected in the mass spectrum (Scheme 4). Even though one equivalent of the Grignard reagent was used, the dialkylated product 17 was admitted as the major product in the mass spectra along with the starting material 7. We postulated that the 1,4-addition of an ethyl group to the unsaturated ester moiety would occur after the alkylation at the acetal moiety proceeded. We then decided to attempt the 1,4-addition reaction for the aminoindanol derivative 20, which would be obtained from the aminoacetal 7 by chemoselective reduction, with ethylmagnesium bromide. The reduction of 7 with LAH or DIBAL-H gave the corresponding diol 19, and Red-Al® in toluene chemoselectively reduced the ester group to give the primary alcohol as a mixture of olefin isomers, while the saturated derivative gave the corresponding primary alcohol.6 Fortunately, we found that the treatment of 7 with NaBH4 in the presence of BF3·OEt2 in THF at 0 °C successfully and quantitatively provided the desired alcohol 20.9 After obtaining 20, we attempted the 1,4-addition reaction.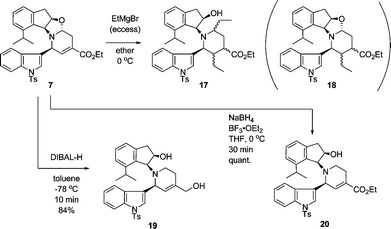 | ||
| Scheme 4 Alkylation and reduction of 7. | ||
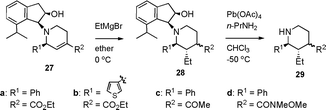
The reaction of 20 with an excess amount of ethylmagnesium bromide in ether at 0 °C for 20 min cleanly proceeded to expectedly afford the 1,4-addition product 21 in 95% yield as a mixture of stereoisomers, which could not be separately isolated at this stage (Scheme 5). Removal of the indanol moiety in 21 with lead tetraacetate under the established condition6 produced the 2,3,4-trisubstituted piperidine 22a and 22b in 24% and 71% yields, respectively, which were cleanly separated from each other. The stereochemistry of 22b was determined as 2β, 3α, 4β in the piperidine ring and that of 22a as 2β, 3α, 4α by a detailed analysis of the NMR data. Although various attempts of isomerization at the ester group of 22a (t-BuOK in BuOH, DBU in DMF, etc.) were not successful, isomerization of the corresponding methyl ketone 5 was successful in literature,3c,d and hence the present synthesis can be regarded as the stereocontrolled one. On the contrary, the reaction of compound 23, which was derived from the hydroxyl compound 20 with TBSCl and imidazole, gave tertiary alcohol 24 with ethylmagnesium bromide resulting from the addition of an ethyl group to the ester group. Thus, the secondary hydroxyl group of the aminoindanol moiety is essential for the 1,4-addition reaction.
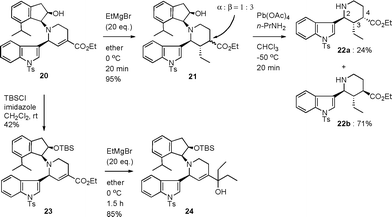 | ||
| Scheme 5 1,4-Addition reaction of unsaturated ester. | ||
A plausible mechanism is shown in Fig. 2. The highly stereoselective introduction of an ethyl group at the β-position of the ester group in 20 could be explained by assuming that the coordination of the Grignard reagent with the OMgBr group prepared from the Grignard reagent and the hydroxyl group in the cis-aminoindanol moiety was generated during the alkylation process,10 and then the ethyl group attacked from the opposite direction of the indole group as shown in structure 26.
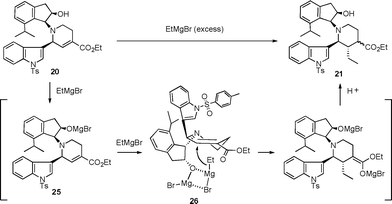 | ||
| Fig. 2 Plausible mechanism of 1,4-addition reaction. | ||
In order to investigate the generality of this novel 1,4-addition reaction assisted by the neighboring hydroxyl group, we applied the reaction to a phenyl derivative 27a instead of an N-tosyl indole compound as a representative compound (Table 1). As expected, the reaction under the same conditions gave the corresponding 1,4-addition product 29a through 28a in 82% yield with the single stereoisomer at the ethyl group and the similar stereoselectivity at the ester group. The thiophene derivative 27b6 gave 29b through 28b in 75% yield and a rather better stereoselectivity at the ester group. On the contrary, attempts of the 1,4-addition reaction of the corresponding methyl ketone derivative 27c, which was easily derived from amide 27d, gave the 1,2-addition product 30c along with 1,4-addition product 29c in 26% and 41% yields, respectively. In the case of the amide derivative 27d, which was obtained by the same one-pot azaelectrocyclization using the corresponding Weinreb amide instead of 14 in Scheme 3 (see experimental), the reaction gave a complex mixture and 29d was not isolated. Thus, the unsaturated ester group was appropriate for the desired 1,4-addition reaction.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 27 | 29 | R1 | R2 | Yield | Ratio α:β |
By-product |
| 1 | 20 | 22 |
|
CO2Et | 95% | 1:3.0 |
|
| 2 | 27a | 29a |
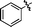
|
CO2Et | 82% | 1:2.7 |
|
| 3 | 27b | 29b |
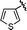
|
CO2Et | 75% | 1:3.8 |
|
| 4 | 27c | 29c |
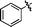
|
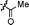
|
41% | 1:2.2 |
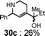
|
 | ||
| Scheme 6 Synthesis of (−)-20-epiuleine. | ||
Total synthesis of (−)-20-epiuleine by the one-pot asymmetric 6π-azaelectrocyclization followed by the novel 1,4-addition
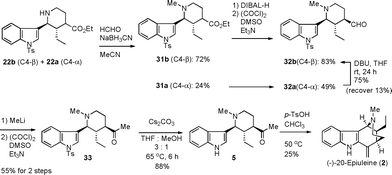
To achieve the total synthesis of 20-epiuleine (Scheme 6), the obtained 22b was transformed into the methyl ketone 5. Thus, the reductive N-methylation of a mixture of 22b and 22a was performed by treatment with a 37% aqueous HCHO solution and sodium cyanoborohydride in acetonitrile to produce 31b and 31a as a mixture of easily separable epimers. Each of them was transformed into the aldehyde 32b and 32a by DIBAL-H reduction followed by Swern oxidation. At this stage, the undesired isomer 32a was epimerized to the desired 32b by a DBU treatment in THF at room temperature. The obtained aldehyde 32b was transformed into the methyl ketone 5 through 33 by methylation with MeLi, Swern oxidation and then hydrolysis of the N-tosyl group of the indole moiety with Cs2CO3 in THF and MeOH (3:1). Finally, the treatment of 5 with p-TsOH in chloroform completed the total synthesis of (−)-20-epiuleine (2) in 25% yield. The spectral data (1H NMR, 13C NMR, IR, HRMS) were in good agreement with those of the natural product. The value of [α]D of the synthesized compound was −30.1 (CHCl3, c 0.9), which is reported for the first time.
Summary
In summary, we achieved the first asymmetric total synthesis of (−)-20-epiuleine (2) using a three-component one-pot 6π-azaelectrocyclization. The construction of the 2,3,4-trisubstituted piperidine as the core structure of the indole alkaloid was established by utilizing the highly stereoselective 1,4-addition reaction only with the Grignard reagent. This unique 1,4-addition reaction was due to the novel neighboring participation of the hydroxyl group of the characteristic aminoindanol moiety, which was employed as a chiral nitrogen source. The total yield of the first route via Husson's enone obtained from the three components in a one-pot reaction was 1.3% for 10 steps, while that of the second route was 5.0% for 11 steps.Experimental
General Procedure
All commercially available reagents were used without further purification. All solvents were used after distillation. Tetrahydrofuran, diethyl ether and toluene were refluxed over and distilled from sodium. Dichloromethane was refluxed over and distilled from P2O5. Dimethylformamide (DMF) was distilled from CaH2. Preparative separation was usually performed by column chromatography on silica gel. The 1H NMR and 13C NMR spectra were recorded using a 400 MHz spectrometer, and chemical shifts were represented as δ-values relative to the internal standard TMS. The IR spectra were recorded by a FT-IR spectrometer. The high-resolution mass spectra (HRMS) were measured by a ESI-TOF MS.:1 mixture of C4 stereoisomers. Column chromatography on silica gel (from 0% to 2.8% methanol in chloroform) gave 22a (159 mg, 24%) as a yellow amorphous solid and 22b (476 mg, 71%) as a yellow amorphous solid. Data for 22a: [α]24D −8.1 (c 0.7, CHCl3); IR (KBr disk, cm−1) 2959, 2934, 2876, 1724, 1370, 1177, 749; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, 1H, J = 8.2 Hz), 7.70–7.75 (m, 3H), 7.54 (s, 1H), 7.29 (dd, 1H, J = 8.2, 7.3 Hz), 7.16–7.23 (m, 3H), 4.39 (d, 1H, J = 9.4 Hz), 4.11–4.25 (m, 2H), 3.13 (ddd, 1H, J = 11.9, 10.1, 3.0 Hz), 2.99 (dd, 1H, J = 8.9, 4.4 Hz), 2.93 (ddd, 1H, J = 11.9, 3.9, 3.9 Hz), 2.32 (s, 1H), 1.78–2.00 (m, 3H), 1.02–1.33 (m, 5H), 0.69 (t, 3H, J = 7.3 Hz); 13C NMR (100 MHz, CDCl3) δ 174.3, 144.7, 135.3, 135.2, 130.3, 130.0, 126.7, 124.6, 123.8, 123.0, 120.7, 113.7, 60.4, 60.0, 53.6, 45.2, 42.5, 40.3, 28.6, 22.5, 21.5, 21.0, 14.3, 14.2, 11.6; ESI HRMSm/z calcd for C25H30N2O4S1 (M+H)+ 455.2005, found 455.1996. Data for 22b: [α]22D −29.1 (c 1.1, CHCl3); IR (KBr disk, cm−1) 2936, 2812, 1728, 1371, 1175, 749; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, 1H, J = 8.2 Hz), 7.79 (d, 1H, J = 7.8 Hz), 7.72 (d, 2H, J = 8.4 Hz), 7.52 (s, 1H), 7.30 (dd, 1H, J = 7.3, 7.1 Hz), 7.22 (dd, 1H, J = 8.2, 7.3 Hz), 7.18 (d, 2H, J = 8.5 Hz), 4.08–4.29 (m, 2H), 3.67 (d, 1H, J = 10.3 Hz), 3.17 (ddd, 1H, J = 11.7, 3.4, 2.7 Hz), 2.76 (ddd, 1H, J = 11.5, 11.2, 3.4 Hz), 2.45 (ddd, 1H, J = 11.2, 11.2, 4.9 Hz), 2.11 (dddd, 1H, J = 10.8, 10.8, 4.1, 4.1 Hz), 1.82–1.96 (m, 2H), 1.26 (t, 3H, J = 7.3 Hz), 1.11–1.21 (m, 1H), 0.97–1.09 (m, 1H), 0.59 (t, 3H, J = 7.6 Hz),; 13C NMR (100 MHz, CDCl3) δ 175.1, 144.8, 135.4, 135.1, 130.0, 126.7, 124.7, 124.3, 123.9, 123.0, 121.0, 113.7; ESI HRMSm/z calcd for C25H30N2O4S1 (M+H)+ 455.2005, found 455.1986.
To a solution of oxalyl chloride (0.162 mL, 5.74 mmol) in dichloromethane (8.0 ml) was added DMSO (0.307 ml, 6.89 mmol) at −78 °C. After the mixture was stirred for 10 min at this temperature, a solution of the produced alcohol (980 mg, 2.30 mmol) in dichloromethane (10.0 ml) was added. After the mixture was stirred for 40 min, triethylamine (1.32 ml, 11.5 mmol) was added. After the mixture was stirred for 5 min at this temperature, then stirred for an additional 20 min at room temperature, H2O was added, and the resulting mixture was extracted with chloroform. The organic layers were combined, washed with a saturated aqueous NaHCO3 solution and brine, dried over MgSO4, filtered and concentrated in vacuo to give the crude products. Column chromatography on silica gel (from 0% to 9.9% methanol in chloroform) gave the aldehyde product 32b (926 mg, 95%) as a yellow amorphous solid: [α]24D −72.4 (c 1.1, CHCl3); IR (KBr disk, cm−1) 2944, 2784, 2705, 1447, 1370, 750; 1H NMR (400 MHz, CDCl3) δ 9.58 (d, 1H, J = 4.1 Hz), 7.99 (d, 1H, J = 8.2 Hz), 7.78–7.86 (br d, 1H, J = 7.3 Hz), 7.70 (d, 2H, J = 8.2 Hz), 7.48 (s, 1H), 7.30 (ddd, 1H, J = 7.3, 7.1, 1.1 Hz), 7.20 (ddd, 1H, J = 7.1, 7.1, 1.0 Hz), 7.16 (d, 2H, J = 8.2 Hz), 3.08 (ddd, 1H, J = 11.5, 3.4, 3.0 Hz), 2.92 (d, 1H, J = 10.3 Hz), 2.26–2.39 (m, 4H), 2.09–2.22 (m, 2H), 1.76–1.93 (m, 5H), 1.07–1.19 (m, 1H), 0.86–0.98 (m, 1H), 0.56 (t, 3H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 204.0, 145.1, 136.0, 135.2, 130.0, 126.9, 125.2, 125.0, 123.5, 123.3, 121.6, 114.2, 65.1, 55.5, 52.3, 44.8, 40.3, 26.1, 23.1, 21.7, 9.5; ESI HRMSm/z calcd for C24H28N2O3S1 (M+H)+ 425.1899, found 425.1902.
To a solution of oxalyl chloride (0.02 mL, 0.77 mmol) in dichloromethane (2.0 ml) was added DMSO (0.04 ml, 0.93 mmol) at −78 °C. After the mixture was stirred for 10 min at this temperature, a solution of the produced alcohol (132 mg, 0.31 mmol) in dichloromethane (2.0 ml) was added. After the mixture was stirred for 40 min, triethylamine (0.18 ml, 1.55 mmol) was added. After the mixture was stirred for 5 min at this temperature, it was stirred for an additional 20 min at room temperature, then H2O was added, and the resulting mixture was extracted with chloroform. The organic layers were combined, washed with a saturated aqueous NaHCO3 solution and brine, dried over MgSO4, filtered and concentrated in vacuo to give the crude products. Column chromatography on silica gel (from 0% to 9.9% methanol in chloroform) gave 32a (83 mg, 63%) as a white amorphous solid: [α]26D −37.1 (c 1.3, CHCl3); IR (KBr disk, cm−1) 2960, 2788, 1717, 1447, 1370, 750; 1H NMR (400 MHz, CDCl3) δ 9.94 (s, 1H), 7.99 (d, 1H, J = 8.5 Hz), 7.78 (d, 1H, J = 7.9 Hz), 7.71 (d, 2H, J = 8.5 Hz), 7.46 (s, 1H), 7.31 (dd, 1H, J = 7.1, 7.1 Hz), 7.15–7.23 (m, 3H) 3.28 (d, 1H, J = 10.3 Hz), 2.15–2.36 (m, 5H), 1.91–2.11 (m, 5H), 1.24–1.39 (m, 1H), 0.87–1.00 (m, 1H), 0.64 (t, 3H, J = 7.3 Hz); 13C NMR (100 MHz, CDCl3) δ 204.5, 144.8, 135.7, 135.0, 129.7, 126.7, 124.9, 124.5, 123.8, 123.1, 121.1, 114.0, 64.3, 52.7, 46.0, 44.3, 43.9, 25.6, 22.3, 21.5, 11.7; ESI HRMSm/z calcd for C24H28N2O3S1 (M+H)+ 425.1899, found 425.1898.
:1 mixture of diastereomers as a yellow amorphous solid: [α]24D −90.6 (c 1.1, CHCl3); IR (KBr disk, cm−1) 2963, 2785, 1447, 1370, 1175, 749; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, 1H, J = 8.4 Hz), 7.76–7.87 (br s, 1H), 7.70 (d, 2H, J = 8.4 Hz), 7.43–7.50 (br s, 1H), 7.25–7.32 (m, 1H), 7.13–7.23 (m, 3H), 4.00–4.10 (m, 1H), 3.04–3.11 (m, 1H), 2.88–2.96 (m, 1H), 3.32 (s, 3H), 1.53–2.20 (m, 8H), 1.08–1.40 (m, 4H), 0.77–0.94 (br m, 1H), 0.57–0.68 (m, 3H);13C NMR (100 MHz, CDCl3) δ 144.7, 144.7, 135.7, 135.0, 129.6, 126.6, 124.7, 124.5, 123.1, 113.9, 113.8, 67.3, 66.4, 56.7, 44.7, 44.6, 43.4, 43.2, 23.4, 23.0, 21.5, 21.0, 20.7, 20.5, 17.2, 8.8, 8.4; ESI HRMSm/z calcd for C25H32N2O3S1 (M+H)+ 441.2212, found 441.2201.
To a solution of oxalyl chloride (0.085 mL, 3.00 mmol) in dichloromethane (3.0 ml) was added DMSO (0.160 ml, 3.60 mmol) at −78 °C. After the mixture was stirred for 20 min at this temperature, a solution of the produced sec-alcohol afforded above (529 mg, 1.20 mmol) in dichloromethane (6.0 ml) was added. After the mixture was stirred for 40 min, triethylamine (0.693 ml, 6.00 mmol) was added. After the mixture was stirred for 5 min at this temperature, it was stirred for an additional 20 min at room temperature, then H2O was added, and the resulting mixture was extracted with chloroform. The organic layers were combined, washed with a saturated aqueous NaHCO3 solution, dried over MgSO4, filtered and concentrated in vacuo to give the crude products. Column chromatography on silica gel (from 0% to 0.5% methanol in chloroform) gave 33 (449 mg, 85%) as a yellow amorphous solid: [α]22D −71.6 (c 1.0, CHCl3); IR (KBr disk, cm−1) 2938, 2784, 1707, 1447, 1340, 1175, 1121; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, 1H, J = 8.2 Hz), 7.78–7.90 (br s, 1H), 7.69 (d, 2H, J = 8.2 Hz), 7.42–7.50 (br s, 1H), 7.30 (ddd, 1H, J = 8.5, 6.1, 1.1 Hz), 7.21 (ddd, 1H, J = 8.0, 7.1, 0.9 Hz), 7.16 (d, 2H, J = 8.0 Hz), 3.06 (ddd, 1H, J = 11.7, 3.4, 3.2 Hz), 2.89 (d, 1H, J = 10.4 Hz), 2.52 (ddd, 1H, J = 11.2, 11.2, 4.8 Hz), 2.30 (s, 1H), 2.07–2.27 (m, 5H), 1.82–1.92 (m, 5H), 0.98–1.06 (m, 1H), 0.74–0.88 (br s, 1H), 0.53 (t, 3H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 211.5, 145.0, 135.9, 135.2, 129.8, 126.8, 125.1, 124.8, 123.5, 123.4, 121.8, 114.1, 56.3, 53.7, 44.7, 29.3, 28.8, 23.1, 21.7, 9.4; ESI HRMSm/z calcd for C25H30N2O3S1 (M+H)+439.2055, found 439.2038.
Acknowledgements
This work was supported by a Grant-in-Aid for Science Research on Innovative Areas and a Matching Fund Subsidy for a Private University.Notes and references
- J. Bosch and J. Bonjoch, Pentacyclic Strychnos Indole Alkaloids. In Studies in Natural Products Chemistry; Atta-ur-Rahman, ed.; Elsevier: Amsterdam, 1988; Vol. 1, pp 31–88 Search PubMed.
- Synthesis of uleine: (a) A. Jackson, N. D. V. Wilson, A. J. Gaskell and J. A. Joule, J. Chem. Soc. C, 1969, 2738 RSC; (b) G. Buchi, S. J. Gould and F. Naf, J. Am. Chem. Soc., 1971, 93, 2492 CrossRef; (c) T. Kametani and T. Suzuki, J. Org. Chem., 1971, 36, 129.
- Synthesis of 20-epiuleine: (a) L. J. Dolby and H. Biere, J. Org. Chem., 1970, 35, 3843 CrossRef CAS; (b) M. Natsume and Y. Kitagawa, Tetrahedron Lett., 1980, 21, 839 CrossRef CAS; (c) M. Harris, R. Besselievre, D. S. Grierson and H.-P. Husson, Tetrahedron Lett., 1981, 22, 331 CrossRef CAS; (d) D. S. Grierson, M. Harris and H.-P. Husson, Tetrahedron, 1983, 39, 3683 CrossRef CAS; (e) E. S. Tasber and R. M. Garbaccio, Tetrahedron Lett., 2003, 44, 9185 CrossRef CAS.
- M. Saito, M. Kawamura, K. Hiroya and K. Ogasawara, Chem. Commun., 1997, 765 RSC . Enantioselective synthesis of Strychnos indole alkaloids and related compounds: J. Bonjoch and D. Sole, Chem. Rev., 2000, 100, 3455 Search PubMed.
- (a) K. Tanaka and S. Katsumura, J. Am. Chem. Soc., 2002, 124, 9660 CrossRef CAS; (b) K. Tanaka, T. Kobayashi, H. Mori and S. Katsumura, J. Org. Chem., 2004, 69, 5906 CrossRef CAS.
- T. Kobayashi, M. Nakashima, T. Hakogi, K. Tanaka and S. Katsumura, Org. Lett., 2006, 8, 3809 CrossRef CAS.
- T. Kobayashi, F. Hasegawa, K. Tanaka and S Katsumura, Org. Lett., 2006, 8, 3813 CrossRef CAS.
- (a) T. Kobayashi, K. Takeuchi, J. Miwa, H. Tsuchikawa and S. Katsumura, Chem. Commun., 2009, 3363 RSC; (b) Y. Li, T. Kobayashi and S. Katsumura, Tetrahedron Lett., 2009, 50, 4482 CrossRef CAS.
- H.-P. Husson and J. Royer, Chem. Soc. Rev., 1999, 28, 383 RSC.
- K. Higashiyama, H. Inoue and H. Takahashi, Tetrahedron, 1994, 50, 1083 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The experimantal details of 27a–d and 29a–c and 1H and 13C NMR spectra of the products. See DOI: 10.1039/c0ob00627k |
| This journal is © The Royal Society of Chemistry 2011 |