Suppression of background sites in molecularly imprinted polymersviaurea-urea monomer aggregation†
Yagang
Zhang
,
Di
Song
,
Julius C.
Brown
and
Ken D.
Shimizu
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, USA. E-mail: shimizu@mail.chem.sc.edu; Fax: +01 803 777 9521; Tel: +01 803 777 6523
First published on 22nd November 2010
Abstract
The molecular imprinting process provides a synthetically efficient route to polymers with tailored recognition properties. However, the binding properties of the templated binding sites are often masked by the more prevalent background binding sites. Therefore, a strategy for reducing the number of background binding sites was developed and evaluated that uses functional monomer aggregation to suppress the formation of background sites. A series of imprinted and non-imprinted polymers was formed using crosslinking urea monomer and were evaluated for their ability to rebind the anionic template, tetrabutylammonium diphenyl phosphate (TBA-DPP). The urea monomer was shown to form linear hydrogen bonded aggregates in solution and in the solid state. Functional monomer aggregation in the polymerization solution was shown to dramatically reduce the numbers of background binding sites by occupying and blocking the urea recognition groups that were not bound to the template molecule. Despite the low aggregation constant of the urea monomer (3.5 M−1 in chloroform), the number of background sites was reduced by more than 60%. We predict that this strategy of using monomers that aggregate to reduce background binding sites is a general one for MIPs and other types of polymers with tailored recognition properties. The key is to identify self-assembling monomers where the guest binding processes are stronger than the aggregation processes.
Introduction
Molecularly imprinted polymers (MIPs) are highly crosslinked polymers formed in the presence of a template molecule. The removal of the template molecule from the crosslinked matrix creates binding sites with shape and functional group complementarity to the template. The molecular imprinting process is an attractive approach for preparing materials with tailored recognition properties because of its synthetic efficiency and low cost.1–4 In many cases, the imprinting process can be carried out in a single vessel using commercially available monomers. Despite their many attractive qualities, the utility of MIPs has been limited by their modest binding properties. Due to the low fidelity of the imprinting process, bindings sites formed by the template make up only a small fraction of the overall number of binding sites.5,6 Instead, the majority of binding sites are background binding sites that possess low affinity and selectivity for the template. One of the major reasons is that a large excess of functional monomer is typically used to ensure the formation of the key monomer·template complex.7–9 A consequence of this strategy is that the majority of functional monomer units are not complexed to a template molecule and form unselective background binding sites (Fig. 1a).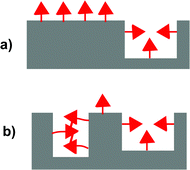 | ||
| Fig. 1 Illustration comparing the relative numbers of background and templated sites in MIPs formed with (a) a functional monomer that does not aggregate and (b) a functional monomer that does aggregate. | ||
The high percentage of background sites in MIPs diminishes their overall affinities and selectivities, and limits their utility in many applications.6,10 Accordingly, a variety of strategies have been developed to improve the ratio of templated to background binding sites. The most direct method has been to design functional monomers with high affinity for the template that can form very strong stoichiometric monomer·template complexes. The stoichiometric imprinting strategy eliminates the necessity to use an excess of functional monomer.11,12 The monomer can also be covalently bonded to the template, ensuring an efficient formation of templated binding sites.3,5 Alternatively, the background sites can be selectively eliminated after the polymerization process via site-selective chemical modification of the recognition groups in the backgrounds sites.13 A general drawback of these approaches is that they require additional synthetic steps. Thus, they diminish one of the primary advantages of the imprinting process, which is synthetic efficiency. In this work, we examine a new method of reducing the number of background sites via functional monomer aggregation. This method does not require additional synthetic steps and is broadly applicable, as most functional monomers display some degree of self-association and new functional monomers can be specifically designed to form aggregates.
Our recent studies of organophosphate imprinted polymers suggested that functional monomer aggregation could have beneficial effects.14 Specifically, MIPs prepared with the urea functional monomers had surprisingly low numbers of background binding sites. We hypothesized that this might be due to the aggregation of the urea monomers that are not bound to the template molecules. The aggregates are captured in the polymer matrix and are not able to form background binding sites as their urea recognition groups are occupied by hydrogen bonds to adjacent monomers (Fig. 1b). The potentially beneficial effects of functional monomer aggregation were initially studied using the functional monomer methacrylic acid, which has the ability to form hydrogen bonded dimers.15Methacrylic acid dimerization in the prepolymerization solution was found to decrease the concentration of free monomer units and very efficiently suppress the formation of background sites. Methacrylic acid dimerization also reduced the number of templated sites but to a much lesser extent. Thus, the overall effect is an improvement in the ratio of background to templated binding sites.
In this study, we examined whether functional monomer aggregation has similar beneficial effects as functional monomer dimerization (Fig. 1b). The influence of functional monomer self-association on the imprinting process has not been extensively studied, and the few studies that have been reported have been primarily computational studies.16–19 The reason for this lack of interest can probably be attributed to the potential of functional monomer aggregation to compete with and disrupt the molecular imprinting process. However, we reasoned that the overall impact could be beneficial if the reductions in the numbers of background sites were larger than the reductions in the numbers of templated sites, as was observed in the case of methacrylic acid dimerization. The key would be to select a monomer in which the monomer·template complexation was much stronger than the monomer·monomer aggregation interactions.
Urea functional monomer 1 (FM 1) was selected for this study based on the following reasons (Fig. 2). First, ureas are well known to form linear aggregates via three point intermolecular hydrogen bonding interactions.20–23 Second, the ureas can form even stronger charge enhanced hydrogen bonded complexes with anionic guests such as phosphates.24–26 Third, FM 1 contains two polymerizable vinyl groups that can rigidly anchor the urea group to the crosslinked matrix. This ensures that functional monomers that are aggregated in solution will remain fixed in an aggregated state in the polymer matrix. Finally, urea functional monomers have been successfully used in molecular imprinting of anionic templates such as phosphates and carboxylates.27–30 For example, we have previously demonstrated that urea FM 1 can be used to imprint the anionic phosphate ester template, tetrabutylammonium diphenyl phosphate (TBA-DPP).14 This template is a safe model for phosphate esters that are the hydrolysis products of insecticides, chemical warfare agents, herbicides, plasticizers, and fire retardants. TBA-DPP also has two aromatic chromophores, which allows its uptake to be easily monitored by UV-vis.
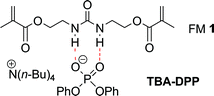 | ||
| Fig. 2 Structure of the hydrogen bonded complex between urea functional monomer 1 and the anionic template TBA-DPP. | ||
Experimental
Chemicals and apparatus
Solvents were purchased from Sigma–Aldrich, Fisher and VWR, and were purified and dried by passing through a PURE SOLV® solvent purification system (Innovative Technology). Deuterated solvents were purchased from Cambridge Isotope Laboratories. All other reagents were purchased from Sigma–Aldrich and were used as received. 1H NMR spectra were recorded on a Varian 300 MHz NMR at ambient temperature. Chemical shifts (ppm) were referenced to tetramethylsilane or residual protonated solvent. UV measurements were made using a Jasco V-530 spectrometer. Gas absorption study was carried out using a Quantachrome Autosorb Automated Gas Sorption System. Surface area and nitrogen adsorption isotherms were calculated by the Brunauer–Emmett–Teller (BET) model.Preparation of FM 1 and TBA-DPP
FM 1 and TBA-DPP were synthesized as previously described.14 In brief, FM 1 was synthesized from the condensation of 2-isocyanatoethyl methacrylate and 2-aminoethyl methacrylate (Scheme 1). TBA-DPP was prepared by deprotonation of diphenyl phosphate with tetrabutyl ammonium hydroxide.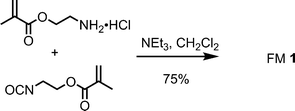 | ||
| Scheme 1 Synthesis of FM 1. | ||
Synthesis of MIPs and NIPs
A series of MIPs and NIPs (Table 1) was prepared in chloroform containing increasing concentrations of DMSO (0% to 45% v/v). The polymers were all formed using similar concentrations of monomers, crosslinkers, and radical initiator.The synthesis of a representative TBA-DPP imprinted polymer proceeds as follows: TBA-DPP (0.098 g, 0.20 mmol), FM 1 (0.171 g, 0.60 mmol), EGDMA (0.754 mL, 4.0 mmol) and AIBN (0.016 g, 0.10 mmol) were dissolved in 2.0 mL of chloroform in a screw-capped vial. The mixture was degassed in an ultrasonic bath for 5 min under nitrogen. The vial was sealed then immersed in a water bath at 65 °C for 6 h. The resulting monolith was crushed and ground with a mortar and pestle. The template and the unreacted species were removed by Soxhlet extraction with methanol for 24 h and then, with a mixture of methanol–acetonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) for another 24 h. The washed MIP particles were dried overnight under vacuum. A non-imprinted control polymer (NIP) was synthesized following the same protocol but without template.
:4 v/v) for another 24 h. The washed MIP particles were dried overnight under vacuum. A non-imprinted control polymer (NIP) was synthesized following the same protocol but without template.
Binding experiments
:1 binding model.31
:1 binding model to yield a Ka = 32 M−1.
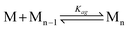 | (1) |
The aggregation constants for urea functional monomers were estimated by fitting the chemical shifts of the N–H protons to the isodesmic equation below using a numerical curve fitting procedure.
| KagCT = (P − Pα)(Pξ − Pα)/(Pξ − P)2 | (2) |
Where Pα and Pξ are the chemical shifts for the free monomers and monomers in the aggregates, P is the observed chemical shift, Kag is the aggregation constant, and CT is total molar concentration of the functional monomers. This analysis yields a Kag of 3.5 M−1 for FM 1 in CDCl3.
Gas adsorption porosimetry
The washed and dried polymers (100 mg) were degassed for 20 h at 25 °C and analyzed by nitrogen adsorption porosimetry (Quantachrome Autosorb Automated Gas Sorption System) at 77.35 K. Surface areas were obtained by the Brunauer–Emmett–Teller (BET) analysis of the 7 point adsorption isotherms and pore size distributions were determined by the Barrett–Joyner–Halenda (BJH) method.Results and discussion
The overall experimental strategy was to: (1) characterize the aggregation and complexation behavior of FM 1 in solution and (2) assess the influences of these association processes on the resulting polymers by comparison of their binding capacities. A series of MIPs and non-imprinted polymers (NIPs) was prepared in chloroform solutions containing increasing amounts of DMSO. The polar DMSO disrupts the aggregation and association processes in the prepolymerization solutions to varying extents depending upon its concentration. Thus, a comparison of the polymer binding capacities provides insight into the importance of these processes in the formation of background and templated binding sites. Particularly insightful were studies of the NIPs (Fig. 3), as the NIPs contain only background binding sites. Thus, a comparison of NIPs formed in solutions with varying DMSO concentrations would yield a direct measure of the relationship between monomer aggregation and the numbers of background binding sites. Specifically, we predicted and found that NIPs formed under conditions where the FM could aggregate would have much lower binding capacities (Fig. 3b).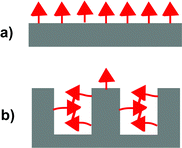 | ||
| Fig. 3 Illustrations of the non-imprinted polymers (NIPs) formed under conditions where the monomers (a) do not aggregate and (b) do aggregate. | ||
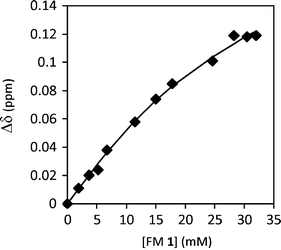
First, the aggregation behavior of urea FM 1 was characterized in solution. The 1H NMR spectra of FM 1 in CDCl3 was measured over a concentration range of 0.35 to 75 mM. The chemical shift of the urea –NHs shifted 0.4 ppm downfield with increasing concentration from 4.61 ppm to 5.02 ppm (Fig. 4), which is consistent with the formation of hydrogen bonded urea aggregates. An aggregation constant of Kag = 3.5 M−1 was obtained by fitting the concentration dependent chemical shift to an isodesmic aggregation model.33 While this study verified the ability of FM 1 to form aggregates, the magnitude of Kag was lower than expected based on comparisons to similar urea aggregation constants reported in the literature.34–36 This could be due to the presence of the four ester oxygens in FM 1 that can form intramolecular 5- and 7-membered hydrogen bonds to the adjacent urea NH’s. In addition, FM 1 is an aliphatic urea, which is less acidic than the aromatic ureas found in most receptors.
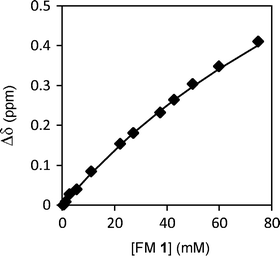 | ||
| Fig. 4 1H NMR chemical shift of urea N–H protons for FM 1 measured from 0.35 mM to 75 mM in CDCl3, which were curve fit to an isodesmic aggregation model. | ||
The ability of DMSO to disrupt FM 1 aggregation was verified by measuring the 1H NMR spectra of FM 1 over a range of concentrations in DMSO-d6. In contrast to the dilution experiment in chloroform, the chemical shift of the urea N–H's remained constant at 6.1 ppm over a similar concentration range (1.5 mM to 50 mM). This is consistent with the ability of DMSO to disrupt urea–urea aggregation by forming strong hydrogen bond interactions to the urea N–H's. The presence of these DMSO–urea interactions could be seen from the much more downfield chemical shifts of the N–H's in DMSO-d6 (6.1 ppm) versus in CDCl3 (<5.1 ppm).
The association processes between FM 1 and the anionic template, TBA-DPP, were similarly characterized by a 1H NMR titration in CDCl3 (Fig. 5). Upon addition of TBA-DPP, the N–H protons of FM 1 shifted 1.2 ppm downfield (4.61 to 5.81 ppm), which was three times larger than the shift observed during the aggregation study and was indicative of a strong association process. The titration data was curve fit to a 1:1 binding model37 to yield a Ka of 176 M−1 for the FM 1·(TBA-DPP) complex.38,39 The magnitude of the binding constant value was comparable to other urea monomer–anion association constants reported in the literature.40–43 More importantly, the FM 1·(TBA-DPP) association constant was much larger than the FM 1 aggregation constant. Thus, the templation processes can out-compete the aggregation processes, enabling the formation of the monomer–template complex in the prepolymerization solution.
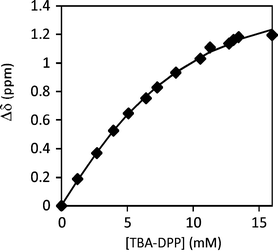 | ||
| Fig. 5
1H NMR titration curve of the addition of 20 mM TBA-DPP to 6 mM FM 1 in CDCl3. The measured chemical shifts of FM 1urea –NH protons were fitted to a 1:1 binding model to yield a Ka = 176 M−1. | ||
The aggregation behavior of FM 1 was also characterized in the solid state. FM 1 formed needle-like crystals on slow evaporation from CH2Cl2. X-Ray crystallographic analysis revealed that FM 1 formed linear hydrogen bonded aggregates (Fig. 6). The individual monomers were held together by intermolecular three-centered urea –NH⋯O hydrogen bonds. The urea –NH⋯O distances of 2.09 Å, and the H⋯O⋯H angle of 61.04° fall into the typical range for urea aggregates (1.98 to 2.21 Å).23
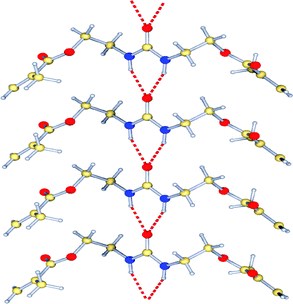 | ||
| Fig. 6 X-Ray crystal structure of FM 1, showing the linear hydrogen bonded aggregates. | ||
In the second set of studies, the effects of FM aggregation on the binding properties of the resulting polymers were studied by systematically disrupting the association processes in the prepolymerization solution. First, a representative MIP and NIP were prepared in order to establish the ability of FM 1 to effectively imprint TBA-DPP. The polymers were prepared in chloroform, using urea 1 as functional monomer, TBA-DPP as template, and ethylene glycol dimethacrylate (EGDMA) as crosslinker. The polymers were synthesized under thermally initiated free radical polymerization conditions (2% AIBN, 65 °C, 6 h). A high 3:1 FM to template ratio was used to ensure the formation of the key monomer·template complexes. The polymers were ground to a fine powder, washed by Soxhlet extraction to remove the template, and dried in vacuo. The binding capacities of the polymers were measured by batch binding studies in which the polymer (60 mg) was equilibrated in a 0.5 mM solution of TBA-DPP (3.5 mL) in CHCl3. After filtration to remove the polymer particles, the change in absorbance of the solution (266 nm) was used to measure change in the free TBA-DPP concentration. While this measurement does not provide the absolute binding capacities of these polymers, the uptake studies do provide a measure of their relative binding capacities. A strong imprinting effect was observed, as the MIP (23 μmol g−1) displayed a four times higher uptake capacity than the NIP (5.4 μmol g−1) (Fig. 7, polymers 1 and 3).
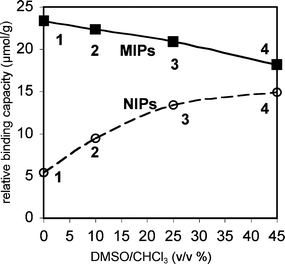 | ||
| Fig. 7 Relative binding capacities for TBA-DPP of MIPs (filled squares) and NIPs (open circles) prepared in solutions of increasing polarity from pure CHCl3 to 45% v/v DMSO–CHCl3, as measured by uptake studies using 60 mg polymer in 3.5 mL of 0.5 mM TBA-DPP in CHCl3. The error for the uptake studies was ±1 μmol g−1. | ||
Next, polymers were prepared in which the association and aggregation processes were systematically disrupted by the addition of DMSO to the prepolymerization solutions (0% to 45% v/v). Comparison of the binding capacities of the MIPs and NIPs showed opposing trends (Fig. 7). The binding capacities of the MIPs decreased whereas the NIPs increased with increasing percentages of DMSO. The modest decrease in relative binding capacity of the MIPs from 23 to 18 μmol g−1 was consistent with current models of the imprinting process.44,45 The polar DMSO disrupts the formation of the FM·template complexes, leading to a decrease in the number of templated sites and in the overall binding capacity. In contrast, simple imprinting models could not explain the dramatic three-fold increase in relative binding capacity of the NIPs formed in non-polar and polar solvents from 5.4 to 15 μmol g−1. This observation, however, was consistent with our hypothesis that FM aggregation could suppress the formation of background sites. The low binding capacity of the NIPs formed in pure CHCl3 was due to the high percentages of urea groups that were in aggregates and prevented from forming background sites. The addition of DMSO disrupts FM aggregation, leading to a higher percentage of free urea groups and a larger number of background sites. Perhaps the most surprising aspect of this study was the magnitude of the effect considering the low aggregation constant of FM 1. Using the values from the uptake studies as an estimate of the number of background sites, the aggregation of FM 1 in chloroform reduced the number of potential background sites by two-thirds.
To confirm that the changes in binding capacity were due to functional monomer aggregation, the polymer morphology was characterized in order to rule out the possibility that differences in polymer surface might be responsible for the observed trends. The surface areas and average pore diameters of the representative MIPs and NIPs were calculated from the nitrogen adsorption isotherms of representative polymers (Table 2). The results show that multi-point BET surface areas and average pore diameters do not explain the observed changes in the relative binding capacities. For example, NIP-1 formed in pure CHCl3 and NIP-4 formed in 45% v/v DMSO–CHCl3 had nearly identical surface areas (330 and 320 m2 g−1) and average pore diameters (55 and 57 Å). This is in contrast to the three-fold difference in binding capacities of NIP-1 and NIP-4. The MIP-1 and MIP-4 formed in non-polar and polar solvent environments did have different surface areas. However, there was no correlation between the binding capacities and the surface areas of the MIPs. MIP-1 with the highest binding capacity has a significantly lower surface area than MIP-4. These results suggested that the differences in binding capacity were not due to differences in surface areas of the polymers and were instead most likely due to the ability of DMSO to disrupt the aggregation and complexation processes of FM 1.
| Polymer | Polymerization solvent | Surface area/m2 g−1 | Average pore diameter/Å |
|---|---|---|---|
| MIP-1 | CHCl3 | 150 | 59 |
| MIP-4 | 45% DMSO/CHCl3 | 280 | 65 |
| NIP-1 | CHCl3 | 330 | 55 |
| NIP-4 | 45% DMSO/CHCl3 | 320 | 57 |
A key question was how could FM aggregation be so effective in suppressing the formation of background sites with such a low FM aggregation constant (Kag = 3.5 M−1)? The first answer is that a high concentration of FM 1 (>170 mM) was used in the imprinting process, which off-sets the low aggregation constant. From this concentration and the measured FM 1 aggregation constant (3.5 M−1), we can estimate that one-third (33%) of the FM 1 was aggregated. However, this explains only one-half of the observed effect, as comparison of the binding capacities of NIP-1 and NIP-4 suggests that two-thirds of the urea groups are aggregated in NIP-1. The second contributing factor may be the higher aggregation constants of the oligomers that are formed during the imprinting process. The polymerization process is not instantaneous and proceeds over a period of hours. Thus, oligomers of FM 1 and EGDMA are present early in the polymerization process. These oligomers contain multiple FM 1 units and thus should have higher aggregation constants, leading to percentages of urea groups participating in urea–urea interactions.
How quickly the relative binding capacities of the MIPs and NIPs converge in Fig. 7 provides a measure of the strengths of the templation and aggregation processes. The convergence point, where the MIP and NIP have the same binding capacity, represents the percentage of DMSO required to completely disrupt both the templation and the aggregation processes. Surprisingly, the convergence point was not reached in this study even with the addition of 45% v/v DMSO to the polymerization mixture. Closer examination of the shape of the curves in Fig. 7 suggests that the lack of convergence is primarily due to the ability of FM1 to still bind and imprint the template in the very polar solvent system. This lead to a modest imprinting effect and higher relative binding capacity for the MIP. The more polar solvent mixture, on the other hand, appears to have completely disrupted the FM 1 aggregation processes in the NIP. This can be seen by the observation that the relative binding capacities of the NIPs in Fig. 7 appear to reach their asymptotic value in the most polar solvent system.
To confirm this analysis, the FM 1 aggregation and FM 1·template binding constants were measured in 45% v/v DMSO–CHCl3. No FM 1 aggregation was observed as the 1H NMR chemical shift of the urea N–H remained constant over a wide concentration range in 45% v/v DMSO-d6–CDCl3. In contrast, significant FM 1·template association was observed, which was consistent with our hypothesis that there is a residual imprinting effect in polymerization solutions with high percentages of DMSO. The titration of FM 1 with TBA-DPP in 45% v/v DMSO-d6–CDCl3 yielded a Ka of 32 M−1 (Fig. 8). While the Ka was lower than in pure chloroform, it is sufficient to yield a modest imprinting effect in 45% v/v DMSO–CHCl3.
 | ||
| Fig. 8
1H NMR titration curve of the addition of TBA-DPP to FM 1 in 45% DMSO-d6–CDCl3. The measured chemical shifts of FM 1urea –NH protons were fitted to a 1:1 binding model to yield a Ka = 32 M−1. | ||
This study demonstrates that one must be very careful when using NIPs as control systems to characterize the imprinting effect. For example, it would be very easy to conclude that the three-fold difference in relative binding capacity of the NIPs formed in non-polar and polar solvents is due to an imprinting effect. Instead, the difference in binding capacity appears to be due to the ability of DMSO to disrupt monomer aggregation. Thus, characterization of the imprinting effect solely by the differences in binding capacity of a MIP and NIP can lead to the inaccurate assignment or overestimation of the imprinting effect. In the case of the TBA-DPP MIP and NIP, approximately half of the difference in binding capacity appears to be due to the ability of TBA-DPP to suppress FM 1 aggregation, and only half of the difference appears to be due to the templation effect. In fact, the presence of an imprinting effect, in this system, was only confirmed by the observed decrease in binding capacity of the MIPs formed in more polar solvents, as only the disruption of the template effect in the original MIP could explain this decrease in binding capacity.
Conclusions
This study of TBA-DPP imprinted urea MIPs and NIPs showed that functional monomer aggregation can greatly reduce the number of background sites by blocking and occupying recognition groups that are not bonded to template molecules. The effects of FM aggregation were clearly observed by comparison of the relative binding capacities of the NIPs formed in polar versusnon-polar solvents, as the polar solvents disrupt the aggregation process in the NIP prepolymerization mixture. The large increase in binding capacity of the NIPs formed in polar solvents showed that a high percentage of the monomers in the NIP formed in the non-polar solvents, such as those used in imprinting, were aggregated and prevented from forming background binding sites. Functional monomers that aggregate can still form imprinted sites. The key is to identify functional monomers and template pairings, which form strong complexes, which can out-compete the self-association processes. We predict that functional monomer aggregation can improve imprinting efficiency in terms of selectivity by suppressing formation of background binding sites. This strategy is orthogonal to the other methods and can be used in conjunction with them to reduce the numbers of background binding sites. Finally, an important consequence of functional monomer aggregation was that it complicates the evaluation of the imprinting effect simply by comparison of the differences in capacity of a MIP and NIP. This study highlights the necessity to use complementary methods to verify and characterize the imprinting effect.Acknowledgements
The authors thank the National Science Foundation (CBET 0828897) for financial support.Notes and references
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- S. C. Zimmerman and N. G. Lemcoff, Chem. Commun., 2004, 5–14 RSC.
- G. Wulff, Chem. Rev., 2002, 102, 1–27 CrossRef CAS.
- M. Yan and O. Ramstrom, eds, Molecularly Imprinted Materials: Science and Technology, Marcel Dekker, New York, 2005 Search PubMed.
- R. J. Umpleby, II, M. Bode and K. D. Shimizu, Analyst, 2000, 125, 1261–1265 RSC.
- P. Szabelski, K. Kaczmarski, A. Cavazzini, Y. B. Chen, B. Sellergren and G. Guiochon, J. Chromatogr., A, 2002, 964, 99–111 CrossRef CAS.
- H. S. Andersson, J. G. Karlsson, S. A. Piletsky, A. C. Koch-Schmidt, K. Mosbach and I. A. Nicholls, J. Chromatogr., A, 1999, 848, 39–49 CrossRef CAS.
- R. J. Ansell and K. L. Kuah, Analyst, 2005, 130, 179–187 RSC.
- R. J. Umpleby, II, A. M. Rampey, S. C. Baxter, G. T. Rushton, R. N. Shah, J. C. Bradshaw and K. D. Shimizu, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 804, 141–149 CrossRef CAS.
- A. M. Rampey, R. J. Umpleby, G. T. Rushton, J. C. Iseman, R. N. Shah and K. D. Shimizu, Anal. Chem., 2004, 76, 1123–1133 CrossRef CAS.
- G. Wulff and K. Knorr, Bioseparation, 2001, 10, 257–276 CrossRef CAS.
- J. L. Urraca, A. J. Hall, M. C. Moreno-Bondi and B. Sellergren, Angew. Chem., Int. Ed., 2006, 45, 5158–5161 CrossRef CAS.
- R. J. Umpleby, G. T. Rushton, R. N. Shah, A. M. Rampey, J. C. Bradshaw, J. K. Berch and K. D. Shimizu, Macromolecules, 2001, 34, 8446–8452 CrossRef.
- X. G. Wu, K. Goswami and K. D. Shimizu, J. Mol. Recognit., 2008, 21, 410–418 CrossRef CAS.
- Y. Zhang, D. Song, L. M. Lanni and K. D. Shimizu, Macromolecules, 2010, 43, 6284–6294 CrossRef CAS.
- L. Malosse, P. Palmas, P. Buvat, D. Ades and A. Siove, Macromolecules, 2008, 41, 7834–7842 CrossRef CAS.
- P. Manesiotis, A. J. Hall and B. Sellergren, J. Org. Chem., 2005, 70, 2729–2738 CrossRef CAS.
- R. J. Ansell, D. Y. Wang and J. K. L. Kuah, Analyst, 2008, 133, 1673–1683 RSC.
- R. J. Ansell and D. Y. Wang, Analyst, 2009, 134, 564–576 RSC.
- J. Yang, M. B. Dewal and L. S. Shimizu, J. Am. Chem. Soc., 2006, 128, 8122–8123 CrossRef CAS.
- S. Boileau, L. Bouteiller, F. Laupretre and F. Lortie, New J. Chem., 2000, 24, 845–848 RSC.
- M. C. Etter, Z. Urbanczyklipkowska, M. Ziaebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- R. Custelcean, Chem. Commun., 2008, 295–307 RSC.
- V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS.
- K. H. Choi and A. D. Hamilton, Coordin Chem Rev, 2003, 240, 101–110 Search PubMed.
- Z. G. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
- J. D. Lee, N. T. Greene, G. T. Rushton, K. D. Shimizu and J. I. Hong, Org. Lett., 2005, 7, 963–966 CrossRef CAS.
- A. Kugimiya and H. Takei, Anal. Chim. Acta, 2006, 564, 179–183 CrossRef CAS.
- A. J. Hall, L. Achilli, P. Manesiotis, M. Quaglia, E. De, Lorenzi and B. Sellergren, J. Org. Chem., 2003, 68, 9132–9135 CrossRef CAS.
- P. Manesiotis, A. J. Hall, M. Emgenbroich, M. Quaglia, E. De, Lorenzi and B. Sellergren, Chem. Commun., 2004, 2278–2279 RSC.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley, New York, 1987 Search PubMed.
- V. Simic, L. Bouteiller and M. Jalabert, J. Am. Chem. Soc., 2003, 125, 13148–13154 CrossRef CAS.
- R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS.
- J. Yang, M. B. Dewal, D. Sobransingh, M. D. Smith, Y. W. Xu and L. S. Shimizu, J. Org. Chem., 2009, 74, 102–110 CrossRef CAS.
- F. Lortie, S. Boileau and L. Bouteiller, Chem.–Eur. J., 2003, 9, 3008–3014 CrossRef CAS.
- M. Barboiu, S. Cerneaux, d. L. van, A and G. Vaughan, J. Am. Chem. Soc., 2004, 126, 3545–3550 CrossRef CAS.
- Previous studies showed that FM 1 forms 1:1 complexes with TBA-DPPref. 14.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- The value of Ka = 176 M−1 for the FM 1·(TBA-DPP) complex is slightly higher than our previously reported value of 133 M−1in ref. 14. The differences appear to be due to the poor solubility of TBA-DPP in CDCl3. The more recent measurement was more accurate as the TBA-DPP concentrations of each point were remeasured and corrected based on the relative areas of the host and guest peaks in the 1H NMR spectra.
- A. Vacca, C. Nativi, M. Cacciarini, R. Pergoli and S. Roelens, J. Am. Chem. Soc., 2004, 126, 16456–16465 CrossRef CAS.
- T. Shioya, S. Nishizawa and N. Teramae, J. Am. Chem. Soc., 1998, 120, 11534–11535 CrossRef CAS.
- H. H. Zepik and S. A. Benner, J. Org. Chem., 1999, 64, 8080–8083 CrossRef CAS.
- E. Fan, S. A. Vanarman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS.
- A. M. Rosengren, J. G. Karlsson, P. O. Andersson and I. A. Nicholls, Anal. Chem., 2005, 77, 5700–5705 CrossRef CAS.
- E. V. Piletska, A. R. Guerreiro, M. J. Whitcombe and S. A. Piletsky, Macromolecules, 2009, 42, 4921–4928 CrossRef CAS.
Footnote |
| † CCDC reference number 791415. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ob00637h |
| This journal is © The Royal Society of Chemistry 2011 |