Practical access to highly enantioenriched quaternary carbon Michael adducts using simple organocatalysts†
Thomas C.
Nugent
*,
Mohammad
Shoaib
and
Amna
Shoaib
Department of Chemistry, School of Engineering and Science, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: t.nugent@jacobs-university.de; Fax: +49 421 200 3229; Tel: +49 421 200 3232
First published on 21st October 2010
Abstract
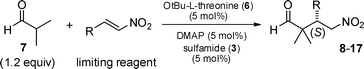
A three component catalyst system entailing an amino acid (OtBu-L-threonine), a hydrogen bond donor (sulfamide), and an amine base (DMAP) allows α-branched aldehyde addition to nitroalkenes in good to high yield and excellent ee. Importantly, the lowest reported catalyst loading (5.0 mol%) and aldehyde stoichiometry (1.2–2.0 equiv) is demonstrated and in most instances the best current product profile is observed.
Natural product and pharmaceutical drug synthesis present multiple levels of challenge and complication that must be concurrently overcome. Beyond the chosen synthetic strategy, a simplistic view holds that construction of the carbon backbone and functional group manipulation are the basic tasks. In this context methods allowing carbon–carbon bond forming reactions under mild conditions are of interest and especially so when they address the long standing problem of stereogenic quaternary carbon bond formation.1,2 The Michael reaction affords this possibility when employing α,α′-substituted aldehydes, and the resulting products would be considered high value multifunctional building blocks for natural product elaboration.
Organocatalysts requiring synthesis are common and reports on their facilitation of unbranchedaldehyde, e.g. 2–3 equiv of n-pentanal, addition to β-nitrostyrene in the presence of 1–5 mol% of a proline derivative allow product formation in excellent yield, dr, and ee.3 However, these low catalyst loadings are not sustainable when branched (α,α- or α,α′-substituted) aldehydes are examined.4 Investigations detailing the use of multiple α,α-substituted and α,α′-substituted aldehydes are uncommon and have waned since the thorough reports of Barbas,5 Jacobsen,6 and Connon.7 These reports and others highlight the continuing challenge associated with quaternary carbon bond formation, and expound the use of 10 mol%,8,9 20 mol%,10,11 or 30 mol%12 organocatalyst loading to add isobutyraldehyde (minimum of 2.0 equiv)13 to 2-substituted-nitroethenes. Hence, advances have been made, but room for improvement remains regarding catalyst loading, aldehyde stoichiometry, substrate scope, and reaction time.
Two bodies of unrelated literature piqued our interest in the Michael reaction. The first is recent, constituting amino acid catalyzed Michael reactions in the presence of a base,10a,d,f,14,15 while the other has been studied for decades and details the now well understood self-assembly of carboxylate anions with hydrogen bond donors, e.g.amino acids with ureas or thioureas.16 Our open question was whether the self-assembly of a hydrogen bond donor to the carboxylate of an amino acid would facilitate cooperative bifunctional organocatalysis, and thereby constructively impact stereoselectivity, reaction rate, and/or substrate breadth. In this light, we demonstrate here what we believe is the first synergistic use of an amino acid, a hydrogen bond donor, and a base for an organocatalytic reaction. Importantly, each catalyst component is commercially available.
Amino acids are well-known to favor their zwitterionic form. In this context addition of a base, to free the primary amine for catalysis, made sense. Our proof of concept succeeded when we combined isoleucine, Et3N, and urea (1, Fig. 1) in the presence of isobutyraldehyde and β- nitrostyrene (Table 1, entry 1).17 The reaction was further optimized (Table 1) by examining a large number of amino acid, hydrogen bond donor, and base combinations (see ESI for a complete list†).18,19 Entries 3, 6, 8, 9, and 12 (Table 1) are of particular interest and five salient points follow. (1) The lowest aldehyde-to-β-nitrostyrene (1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0) ratio is used (entries 4, 6, 9).13 (2) The lowest catalyst loading (OtBu-L-threonine, 2 mol%) to date was demonstrated (entries 3 & 4). (3) High yield and enantioselectivity are coupled with short reaction times (3–7 h).4,8–12,15 (4) The active organocatalyst is extraordinary because it requires no synthesis, requiring only the addition of commercially available compounds (Fig. 1). Finally, (5) to our knowledge this is the first demonstration of the utility of the most simple sulfamide (Fig. 1, 3) as a hydrogen bond donor in the field of organocatalysis.
:1.0) ratio is used (entries 4, 6, 9).13 (2) The lowest catalyst loading (OtBu-L-threonine, 2 mol%) to date was demonstrated (entries 3 & 4). (3) High yield and enantioselectivity are coupled with short reaction times (3–7 h).4,8–12,15 (4) The active organocatalyst is extraordinary because it requires no synthesis, requiring only the addition of commercially available compounds (Fig. 1). Finally, (5) to our knowledge this is the first demonstration of the utility of the most simple sulfamide (Fig. 1, 3) as a hydrogen bond donor in the field of organocatalysis.
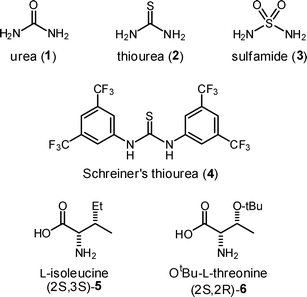 | ||
| Fig. 1 Hydrogen bond donors and amino acids of interest. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 7 (equiv) | L-AA 5 or 6/mol (%) | H-bond donor/mol (%) | DMAP (mol%) | Time/h | Yield (%)b | ee (%)c |
| a 25 °C, toluene (1.0 M), except entries 3–6: cyclohexane (1.0 M) is optimal. b Isolated yield data. c HPLC data (Chiralcel OD-H column). d Et3N was used instead of DMAP. e HPLC area % data. f Preformed lithium salt of OtBu-L-threonine (5 mol%) used, no DMAP added. | |||||||
| 1 | 2 | isoLeu/10 | Urea 1/10 | 10d | 24 | 53 | 90 |
| 2 | 2 | isoLeu/5 | Sulfamide 3/5 | 5 | 24 | 65 | 97 |
| 3 | 2 | OtBu-Thr/2 | Thiourea 4/2 | 8 | 5 | 82 | 93 |
| 4 | 1.2 | OtBu-Thr/2 | Thiourea 4/2 | 8 | 5 | 65 | 91 |
| 5 | 2 | OtBu-Thr/5 | Thiourea 4/5 | 5 | 2 | 99 | 94 |
| 6 | 1.2 | OtBu-Thr/5 | Thiourea 4/5 | 5 | 3 | 99 | 93 |
| 7 | 2 | OtBu-Thr/3 | Sulfamide 3/3 | 3 | 24 | 86 | 97 |
| 8 | 2 | OtBu-Thr/5 | Sulfamide 3/5 | 5 | 4 | 99 | 98 |
| 9 | 1.2 | O t Bu-Thr /5 | Sulfamide 3/5 | 5 | 7 | 97 | 98 |
| 10 | 1.2 | OtBu-Thr/5 | — | 5 | 7 | ∼5e | — |
| 11 | 1.2 | OtBu-Thr/5 | Sulfamide 3/5 | — | 7 | ∼0e | — |
| 12 | 2 | OtBu-Thr (Li salt)/5 | Sulfamide 3/5 | —f | 5 | 99 | 95 |
| 13 | 2 | OtBu-Thr (Li salt)/5 | — | —f | 24 | 92 | 88 |
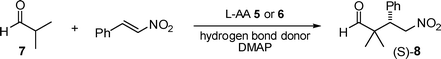
Although thiourea 420 allowed a 2.0 mol% catalyst loading it consistently provided lower enantioinduction than 3 (5 mol%), see Table 1 (entries 3 & 4 vs. 8 & 9). Consequently, we used the sulfamide (3) hydrogen bond donor for the remainder of our study. It is important to note that removal of one of the three catalyst components results in reaction failure (Table 1, entries 9–11). Interestingly, the lithium salt of OtBu-L-threonine enabled the reaction to occur without the presence of a hydrogen bond donor (Table 1, entry 13), but required greater quantities of the aldehyde (2.0 equiv vs. 1.2 equiv), provided notably lower ee (88 vs. 98), and drastically longer reaction time (24 vs. 7 h). These limitations made the lithium salt, alone, impracticable for the more challenging substrates shortly discussed. Furthermore, when using the optimized reaction conditions (Table 1, entry 9) replacing sulfamide (3) by urea (1) or thiourea (2) resulted in much slower reactions and moderate product ee (80%) for both. Finally, n-heptane can be used instead of toluene without detriment.
A current theme within organocatalysis is to meet the stoichiometry requirements governing practical organic synthesis. In this vein, we reacted a range of electron deficient and rich aryl substituted β-nitrostyrenes with only 1.2 equiv of isobutyraldehyde (Table 2, entries 1–8).13 In all instances, except one (Table 2, entry 7), excellent isolated yields 85–98% and ees ≥96% were found within 24 h. Of particular note, 2-isobutyl-nitroethene and 2-styryl-nitroethene were examined to highlight the electronic, steric, and functional group diversity the method can afford. Again using 1.2 equiv of isobutyraldehyde, both substrates provided excellent product profiles (Table 2, entries 9 and 10), in fact the best currently known.7,10d The method is of further value because several products (Table 2, entries 1–4, 8, and 10) could be isolated in high chemical purity (>96%, 1H NMR and HPLC) after a simple water work-up and high vacuum drying. This was possible because these reactions, in general, are by-product free, the β-nitrostyrene was fully consumed, the small excess of volatile isobutyraldhyde was readily removed under high vacuum drying, and the amino acid, DMAP, sulfamide catalyst system is water soluble on work-up.
|
|
||||
|---|---|---|---|---|
| Entry | Product, R = | Time/h | Yield (%)b | ee (%)c |
| a 25 °C, toluene (1.0 M). b Isolated yield data after water work-up followed by high vacuum drying, see ESI data for proof of purity (1H NMR and HPLC).† c HPLC data. d Required chromatography. | ||||
| 1 | C6H5 (8) | 7 | 97 | 98 |
| 2 | p-ClC6H4 (9) | 24 | 98 | 96 |
| 3 | p-FC6H4 (10) | 24 | 93 | 98 |
| 4 | o-BrC6H4 (11) | 24 | 88 | 96 |
| 5 | p-MeC6H4 (12) | 24 | 85d | 99 |
| 6 | p-MeOC6H4 (13) | 24 | 90d | 98 |
| 7 | o-MeOC6H4 (14) | 36 | 72d | 96 |
| 8 | 2-furyl (15) | 8 | 98 | 98 |
| 9e |
|
36 | 70d | 96 |
| 10e |
|
6 | 98 | 97 |
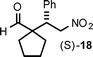
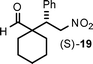
Cyclopentane- and cyclohexanecarboxaldehydes are more sterically congested and infrequently examined. For cyclopentanecarboxaldehyde,5,7,10b,d,e,21 we achieved the top result to date concerning starting material ratio (2:1), catalyst loading (5 mol%), and product profile (Table 3, entry 1). We then turned our attention to cyclohexanecarboxaldehyde which is inexpensive compared to cyclopentanecarboxaldehyde, but essentially not reported on.22,23 The best previous ee was 64%,23 and using our standard 5 mol% catalyst loading with a cyclohexanecarboxaldehyde/β-nitrostyrene ratio of 2:1, we isolated compound (S)-19 in 64% yield with the greatly increased ee of 90% within 30 h. When the catalyst loading was increased to 10 mol% the yield increased to 88% (Table 3, entries 2 and 3).
| Entry | Product | t/h | Yield (%)b | dr c | ee (%)c |
|---|---|---|---|---|---|
|
a Reaction conditions: aldehyde/nitroalkene (2:1), OtBu-L-threonine (6) (5 mol%), sulfamide (3) (5 mol%), DMAP (15 mol%) in toluene (1.0 M) at 25 °C.
b Isolated yield data after chromatography.
c
HPLC analysis.
d
OtBu-L-threonine (6) (10 mol%), sulfamide (3) (10 mol%), DMAP (10 mol%) in toluene (1.0 M) at 25 °C.
|
|||||
| 1 |
|
7 | 89 | — | 97 |
| 2 |
|
30 | 64 | — | 90 |
| 3d | 48 | 88 | — | 91 | |
| 4 |
|
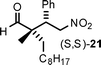
12 | 84 | 70:30 |
97 |
| 5 |
|
12 | 71 | 78:22 |
91 |
| 6 |
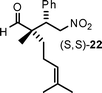
|
12 | 70 | 77:23 |
98 |
| 7 |
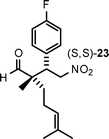
|
36 | 80 | 77:23 |
99 |
| 8 |
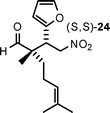
|
16 | 83 | 76:24 |
97 |
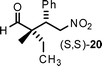
Aldehydes with nonequivalent α-substituents (α,α′-substituted aldehydes) provide Michael products containing stereogenic quaternary carbons and these products were obtained in good yield and excellent ee, but with moderate diastereomeric ratios (Table 3, entries 4–8). The diastereomeric ratios are likely a reflection of the (E)- to (Z)-enamine ratios of the in situ formed enamine. This is supported by the fact that our drs broadly match those reported in the literature for the same (20–22) or similar products (23 and 24) when using unrelated organocatalysts.5–7
Catalyst
self-assembly as a strategy for organocatalytic Michael reactions with β-nitrostyrene is known,15,24 but only Demir has examined quaternary carbon bond formation.25–27 Evidence for catalyst self-assembly in our system is based on precedent and preliminary evidence. For example, it is well established that hydrogen bond donors strongly bind to amino acid carboxylate anions having ammonium counter ions.16 It is therefore a reasonable and strong possibility that sulfamide (3) binds to the carboxylate anion of OtBu-L-threonine having a protonated DMAP counter ion (Fig. 2).28 Furthermore, decreasing the amount of DMAP or sulfamide, relative to the optimal 1:1:1 ratio with OtBu-L-threonine, results in a non-linear decrease in the reaction rate. Moderate increases in the amount of DMAP or sulfamide, relative to the optimal 1:1:1 ratio with OtBu-L-threonine, have little effect on the reaction; but addition of larger quantities of DMAP (15 mol% vs. 5 mol%) results in reaction rate acceleration (Table 3, see reaction conditions). This base effect has been previously described by Yan.10c Finally, the lack of solubility of OtBu-L-threonine and sulfamide, until all three catalyst components are present, is another indicator that self-assembly is occurring. With this background, a preliminary mechanism is proposed.
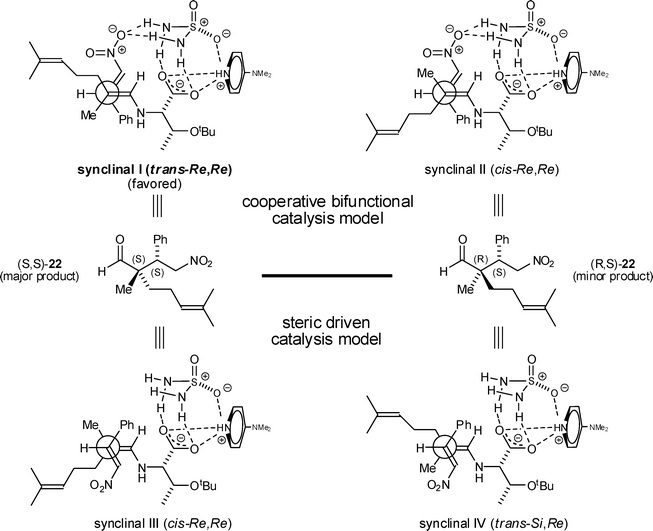 | ||
| Fig. 2 Postulated transition states for α,α′-substituted aldehyde addition to β-nitrostyrene (Table 3, entry 6 modelled). | ||
The presence of a thiourea moiety in a bifunctional Michael organocatalyst is associated with the orbital activation of a nitroalkene and the simultaneous forced proximity (pre-transition organization) of the nitroalkene with the enamine.6,29 Here it is reasonable to assume, after self-assembly has occurred, that a similar scenario unfolds; albeit with sulfamide (3) strongly associating with the carboxylate anion and simultaneously hydrogen bonding with the nitroalkene (Fig. 2, synclinal I).30–32
Why sulfamide (3), and not simple urea (1) or thiourea (2), Fig. 1, is more effective can be rationalized by invoking the distorted tetrahedral geometry at the sulfur atom of 3. This allows one of the oxygens of sulfamide to always be in a position to take part in an electrostatic interaction with the protonated nitrogen atom of DMAP. If occurring, the carboxylate, protonated DMAP, sulfamide aggregate would represent a closed, or cyclic, assembly of the three interacting partners as depicted in Fig. 2. This configuration would be expected to be more rigid and populated than the corresponding ‘open acyclic systems’ based on urea (1) or thiourea (2), leading to increased levels of stereoinduction and increased reaction rate.
When a lithium counter ion is present and no hydrogen bond donor is present (Table 1, entry 13), the basic model would still be valid, but now the nitro group would be directly associated with the lithium cation. To overcome the shortened interaction distance (no hydrogen bond donor intermediary), a synperiplanar transition state (not shown) would be required. In this situation, increased substituent eclipsing effects would raise the transition state energy, slowing down the reaction and possibly allowing the non-productive steric pathway (Fig. 2, synclinal IV) to compete. These after the fact explanations account for the much slower reaction time and lower ee in the absence of a hydrogen bond donor (Table 1, compare entries 9 and 13).
A synclinal Newman transition state model rationalizes the stereochemical outcome of enamine additions to nitroalkenes, as elaborated on in the work of Risaliti33 and Seebach;34 and, more recently, for the amino acid catalyzed version, by Yoshida.10a,f With these points in mind, we sought to explain the observed absolute and relative stereochemistry of our products (8–24). For this purpose, we used an α,α′-substituted aldehyde for our transition state model: 2,6-dimethylhept-5-enal (Table 3, entry 6). The major product is (S,S)-22, based on chiral HPLC, 1H NMR spectroscopic data, and comparison of these data with the published values.5,6 In the proposed transition states (Fig. 2), the L-threonine backbone of the enamine minimizes steric interactions via gauche staggering, which projects the carboxylate moiety to the α-face of the enamine. A reasonable assumption is that the (E)-enamine prevails, in this scenario the synclinal I facial approach accounts for the enantio- and diastereocontrol observed in the major product (S,S)-22 (Fig. 2) and consequently for all other products shown here. The minor product, (R,S)-22, is explainable based on the (Z)-enamine (synclinal II, Fig. 2). A steric based model (synclinal III, no H-bonding assistance) can also explain the stereochemical outcome for the major product (S,S)-22 (Fig. 2). However, it requires the minor (Z)-enamine, and thereby fails to explain how the major product would be formed.
In summary, quaternary carbon–carbon bond formation in these Michael reactions entails cooperative bifunctional catalysis (synclinal I and II, Fig. 2). The amino acid's primary amine transforms the aldehyde into an enamine, while the second functional group, here the carboxylate-sulfamide assembly, activates the nitroalkene and directs it into bonding distance proximity of the β-carbon of the enamineviahydrogen bonding. The carboxylate-sulfamide assembly may also increase the rate of enamine formation by enforcing a conformational preference in the amino acid backbone. These combined facets may account for the low catalyst loading and need for only a small excess of the aldehyde as reported here.
In closing, we have developed a new catalyst assembly based approach for the catalytic enantioselective formation of an important class of Michael products bearing quaternary carbons. Through the hitherto unknown synergistic combination of an amino acid, a base, and a hydrogen bond donor, it is now possible to promote these reactions with lower loadings of both the catalyst and the aldehyde than previously feasible, and within relatively short reaction times. The cooperation of the three catalyst components, and their inherently tunable nature, may open up new avenues for the pursuit of organocatalytic reactions in general.19 Preliminary modelling suggests that other Michael acceptors (e.g.maleimide, DEAD, unsaturated sulfones, etc.), the Mannich reaction, and mechanistically related reactions, will be amenable to this approach.
Acknowledgements
We are grateful to Jacobs University Bremen for funding, MS gratefully acknowledges support from the HEC of Pakistan, and AS gratefully acknowledges a scholarship from TCN (grant number 01).Notes and references
- The Way of Synthesis T. Hudlický and J. W. Reed, ed.; Wiley VCH:Weinheim, 2010, specifically see p. 9 Search PubMed.
- For recent reviews on stereogenic quaternary carbon formation, see: (a) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873–888 RSC; (b) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (c) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473–1482 CrossRef CAS.
- (a) D. Lu, Y. Gong and W. Wang, Adv. Synth. Catal., 2010, 352, 644–650 CrossRef CAS; (b) Z. Zheng, B. L. Perkins and B. Ni, J. Am. Chem. Soc., 2010, 132, 50–51 CrossRef CAS; (c) M. Lombardo, M. Chiarucci, A. Quintavalla and C. Trombinia, Adv. Synth. Catal., 2009, 351, 2801–2806 CrossRef CAS; (d) M. Wiesner, J. D. Revell and H. Wennemers, Angew. Chem., Int. Ed., 2008, 47, 1871–1874 CrossRef CAS; (e) S. Zhu, S. Yu and D. Ma, Angew. Chem., Int. Ed., 2008, 47, 545–548 CrossRef CAS.
- One 5 mol% catalyst loading example was demonstrated, see ref. 3c: isobutyraldehyde/β-nitrostyrene (2:1), 5 mol% catalyst loading, 22 h, 99% yield, 76% ee.
- N. Mase, R. Thayumanavan, F. Tanaka and C. F. Barbas III, Org. Lett., 2004, 6, 2527–2530 CrossRef CAS.
- M. P. Lalonde, Y. Chen and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 6366–6370 CrossRef CAS.
- S. H. McCooey and S. J. Connon, Org. Lett., 2007, 9, 599–602 CrossRef.
- 10 mol% catalyst loading. For lead references of those methods requiring ≤5 equiv of an α,α-substituted aldehyde and providing ≥84% ee, see ref. 3e, 7, and: (a) J. Xiao, F.-X. Xu, Y.-P. Lu and T.-P. Loh, Org. Lett., 2010, 12, 1220–1223 CrossRef CAS; (b) P. Li, L. Wang, M. Wang and Y. Zhang, Eur. J. Org. Chem., 2008, 1157–1160 CrossRef CAS; (c) N. Mase, K. Watanabe, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 4966–4967 CrossRef CAS.
- Regarding 10 mol% catalyst loadings and the lowest reported isobutyraldehyde/β-nitrostyrene ratio (2:1), ref. 3e and 8a provide the best results, respectively: 96 h, 86% yield, 95% ee and 60 h, 97% yield, 92% ee. All other cited work in reference 8 provided < 89% ee.
- 20 mol% catalyst loading. For lead references of those methods requiring ≤ 4 equiv of an α,α-substituted aldehyde and providing ≥85% ee, see ref. 6 and: (a) M. Yoshida, A. Sato and S. Hara, Org. Biomol. Chem., 2010, 8, 3031–3036 RSC; (b) C. Chang, S.-H. Li, R. J. Reddy and K. Chen, Adv. Synth. Catal., 2009, 351, 1273–1278 CrossRef CAS; (c) X.-j. Zhang, S.-p. Liu, J.-h. Lao, G.-j. Du, M. Yan and A. S. C. Chan, Tetrahedron: Asymmetry, 2009, 20, 1451–1458 CrossRef CAS; (d) X.-j. Zhang, S.-p. Liu, X.-m. Li, M. Yan and A. S. C. Chan, Chem. Commun., 2009, 833–835 RSC; (e) Q. Zhang, B. Ni and A. D. Headley, Tetrahedron, 2008, 64, 5091–5097 CrossRef CAS; (f) A. Sato, M. Yoshida and S. Hara, Chem. Commun., 2008, 6242–6244 RSC.
- Ref. 6,10a,c,d,f report ≥98% ee for the reaction of isobutyraldehyde (2.0 to 3.7 equiv, 20 mol% cat. loading) with β-nitrostyrene.
- For a 30 mol% example, see ref. 5.
- To our knowledge the use of < 2.0 equiv of an α,α-branched aldehyde has only been elaborated on by Yoshida, see Table 4 (only entry 2) of ref. 10a.
- (a) L.-q. Gua and G. Zhaoa, Adv. Synth. Catal., 2007, 349, 1629–1632 CrossRef; (b) B. M. Choudary, Ch. V. Rajasekhar, G. G. Krishna and K. R. Reddy, Synth. Commun., 2007, 37, 91–98 CrossRef CAS.
- A single α,α-substituted aldehyde was studied, isobutyraldehyde/β-nitrostyrene (3:1), 5 mol% catalyst loading, 84 h, 71% yield, 85% ee, see Supp Info (page S-6†) of: T. Mandal and C.-G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714–7717 Search PubMed.
- For example, see: (a) M. Hernández-Rodríguez and E. Juaristi, Tetrahedron, 2007, 63, 7673–7678 CrossRef CAS; (b) G. M. Kyne, M. E. Light, M. B. Hursthouse, J. de Mendoza and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2001, 1258–1263 RSC; (c) B. C. Hamann, N. R. Branda and J. Rebek, Jr., Tetrahedron Lett., 1993, 34, 6837–6840 CrossRef CAS.
- The preformed lithium salt of isoleucine/urea (5:5 or 10:10 mol%) resulted overwhelmingly in by-product formation.
- Squaramide is an interesting new hydrogen bond donor, the parent primary diamine would be interesting to examine, see: J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 Search PubMed.
- Interesting to examine will be: amino acids related to isoleucine and L-threonine, alternative O-protected threonines, and β-amino acids. It can be further imagined that replacing the carboxylic acid moiety of amino acids with a sulfate or phosphate ester, or a phosphonate or sulfonate moiety will be valuable to pursue.
- M. Kotke and P. R. Schreiner, (Thio)urea Organocatalysts. In Hydrogen Bonding in Organic Synthesis, P. M. Pihko, Ed.; Wiley-VCH: Weinhein, 2009, p. 141–352 Search PubMed.
- J. Wang, H. Li, B. Lou, L. Zu, H. Guo and W. Wang, Chem.–Eur. J., 2006, 12, 4321–4332 CrossRef CAS.
- See ref. 5: cyclohexylcarboxaldehyde/β-nitrostyrene (2:1), 30 mol% loading, 96 h, 90% yield, 59% ee.
- See ref. 21: cyclohexylcarboxaldehyde/β-nitrostyrene (10:1), 20 mol% loading, 96 h, 42% yield, 64% ee.
- (a) D.-Q. Xu, H.-D. Yue, S.-P. Luo, A.-B. Xia, S. Zhang and Z.-Y. Xu, Org. Biomol. Chem., 2008, 6, 2054–2057 RSC; (b) Z.-B. Li, S.-P. Luo, Y. Guo, A.-B. Xia and D.-Q. Xu, Org. Biomol. Chem., 2010, 8, 2505–2508 RSC.
- To see all of Demir's relevant research, see: (a) A. S. Demir and S. Eymur, Tetrahedron: Asymmetry, 2010, 21, 405–409 CrossRef CAS; (b) A. S. Demir and S. Eymur, Tetrahedron: Asymmetry, 2010, 21, 112–115 CrossRef CAS; (c) Ö. Reis, S. Eymur, B. Reis and A. S. Demir, Chem. Commun., 2009, 1088–1090 RSC.
- Demir's quaternary carbon bond formation conditions and results: isobutyraldehyde/β-nitrostyrene (3:1), 20 mol% proline/20 mol% Schreiner's thiourea, 36 h, 66% yield, 72% ee, as found in ref. 25b.
- Related self-assembly based approaches characterize other reactions, for an aldol reaction and an α-hydroxylation of aldehydes, see respectively: (a) X. Company, G. Valero, L. Crovetto, A. Moyano and R. Rios, Chem.–Eur. J., 2009, 15, 6564–6568 CrossRef; (b) S. L. Poe, A. R. Bogdan, B. P. Mason, J. L. Steinbacher, S. M. Opalka and D. T. McQuade, J. Org. Chem., 2009, 74, 1574–1580 CrossRef CAS.
- (a) Sulfonamides and sulfamides are already established as strong hydrogen bond donors, see: A. A. Rodriguez, H. Yoo, J. W. Ziller and K. J. Shea, Tetrahedron Lett., 2009, 50, 6830–6833 Search PubMed; (b) O. Mammoliti, S. Allasia, S. Dixon and J. D. Kilburn, Tetrahedron, 2009, 65, 2184–2195 CrossRef CAS.
- See for example: (a) C. G. Kokotosa and G. Kokotos, Adv. Synth. Catal., 2009, 351, 1355–1362 CrossRef CAS; (b) D. A. Yalalov, S. B. Tsogoeva and S. Schmatz, Adv. Synth. Catal., 2006, 348, 826–832 CrossRef CAS.
- Depictions of nitro groups hydrogen bonding to thioureas are normally drawn with both N–H groups of the thiourea and both oxygen atoms of the nitro group coplanar and hydrogen bonded, for example see: ref. 20 and ref. 31 Some debate remains in the literature, with a 2006 computational study suggesting a ‘one oxygen model’ (see ref. 29b) and other groups having variously used this model (see ref. 6). Our depicted and favored transition state model, synclinical I (Fig. 2), can only accommodate a one oxygen activation model.
- S. J. Connon, Chem.–Eur. J., 2006, 12, 5418–5427 CrossRef.
- Orbital activation of the nitroalkene alone, e.g. a nitroalkene, sulfamide pair independent of the carboxylate moiety is unlikely. This is borne out by performing the reaction in the absence of a base; the nitroalkene, sulfamide pair would still exist, yet no product formation was noted (Table 1, entry 11).
- (a) A. Risaliti, M. Forchiassin and E. Valentin, Tetrahedron, 1968, 24, 1889–1898 CrossRef CAS; (b) F. P. Colonna, E. Valentin, G. Pittaco and A. Risaliti, Tetrahedron, 1973, 29, 3011–3017 CrossRef CAS; (c) E. Valentin, G. Pittaco, F. P. Colonna and A. Risaliti, Tetrahedron, 1974, 30, 2741–2746 CrossRef CAS; (d) S. Fabrissin, S. Fatutta and A. Risaliti, J. Chem. Soc., Perkin Trans. 1, 1981, 109–112 RSC.
- D. Seebach and J. Goliński, Helv. Chim. Acta, 1981, 64, 1413–1423 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full spectroscopic data for compounds, and copies of 1H NMR spectra and HPLC data. See DOI: 10.1039/c0ob00822b |
| This journal is © The Royal Society of Chemistry 2011 |