Sequential click synthesis of hyperbranched polymersvia the A2 + CB2 approach†
Jin
Han
,
Bo
Zhao
,
Yanqin
Gao
,
Aijin
Tang
and
Chao
Gao
*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, P. R. China. E-mail: chaogao@zju.edu.cn
First published on 29th July 2011
Abstract
Sequential thiol–ene and thiol–yne click chemistry via the A2 + CB2 protocol is presented for simple, rapid, and scalable production of hyperbranched polymers, affording hyperbranched polythioether–ynes with high molecular weight and high degree of branching.
Hyperbranched polymers (HPs) possess unique structural attributes as well as special chemical and physical properties, enabling them to be used in coating, processing additives, nanocarriers for dyes and drugs, nanotemplates for metal catalysts, and so on.1 These tremendous applications are urgently calling for HP synthesis developing towards rapidness, mildness, high-yield and scale-up.2 Several synthesis strategies have been developed. Unlike conventional single-monomer (AB2) and symmetric monomer pair (A2 + B3) strategies, the newly developed couple-monomer methodology (CMM) including A2 + CB2, A2 + BB′2, AB + CD2, and A* + CB2 approaches employs asymmetric monomer pair that can mainly in situ form AB2-type intermediate for polymerization instead of purified AB2 or A2 + B3 monomer, saves much effort on precursor purification and allows one-pot simple acquisition of HPs with low gelation risk.2,3 However, the utilized conventional reactions are relatively slow,3 posing a challenge of how to rapidly prepare HPs while keeping the advantages of CMM.
Herein, we represent a sequential click chemistry (SCC) approach to fast production of HPs from commercial monomer pairs.4 Click chemistry has been widely demonstrated as a versatile tool for construction of complex macromolecules since its first definition in 2004 by Sharpless et al.5 Hence, our synthesis approach holds both merits of CMM and click chemistry: simplicity, low risk of gelation, scalability, fastness, mildness, and modularity. Notably, single click chemistry such as thiol–yne or azide–alkyne reaction has been used for the preparation of HPs in the final polymerization step,6,7 whereas no report has been published to employ click chemistry in all of the reaction steps including the synthesis of precursor and formation of HPs.
As shown in Scheme 1, di-thiols and propargyl acrylate are selected as A2 and CB2 monomers, respectively. In the presence of triethylamine (Et3N), a thiol group of di-thiol can react with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of propargyl acrylateviaMichael addition thiol–ene click chemistry,8,9 mainly affording an AB2-type intermediate of “thiol–alkyne”; the following thiol–yne click polymerization yields HP.6,8
C bond of propargyl acrylateviaMichael addition thiol–ene click chemistry,8,9 mainly affording an AB2-type intermediate of “thiol–alkyne”; the following thiol–yne click polymerization yields HP.6,8
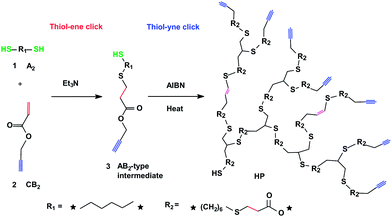 | ||
| Scheme 1 Sequential thiol–ene and thiol–yne click chemistry approach to rapid, easy synthesis of HPs via the A2 + CB2 framework. | ||
Clearly, the AB2-type intermediate is in situ formed in our approach and subsequently used for direct polymerization without further isolation of AB2 monomer. Thus, our approach is essentially different from the classic AB2 approach that needs the synthesis and purification of precursor through conventional reaction with a low efficiency (e.g., 13%) even though thiol–yne click polymerization could be used in the final step.6 Moreover, we employ thermal initiating rather than the generally used UV-light irradiation for the thiol–yne polymerization in order to make the process more straightforward and easily accessible in a large-scale.
Typically, propargyl acrylate 2 was added dropwise to a THF solution of 1,6-hexanedithiol 1 under N2 in the presence of Et3N as a catalyst. After reaction at 35 °C for 6 h till the reaction completion, THF and Et3N were removed under reduced pressure. Toluene containing 2,2′-azobisisobutyronitrile (AIBN) initiator was added subsequently. Further thermo-initiated thiol–yne polymerization was conducted at 65 °C for given times. The whole process was accomplished within 12 h, declaring the high efficiency and cost effectiveness of our new approach indeed.
1H NMR spectra of precursor and HPs are shown in Fig. 1. 13C NMR spectra of precursor and HP are shown in Fig. S1 and S3†, respectively. In the 1H NMR spectrum of precursor, the proton signals at 5.8–6.6 ppm ascribed to the CH2CH moiety of acrylate totally vanished due to the thiol–ene addition,10 new signals at 2.6–2.8 ppm assigned to the resulted CH2CH2 bonds emerged and the ratio of the integration of the proton signals labeled with “b” to the integration sum of the proton signals labeled with “f” and “i” was 1:2, indicating that the precursor was successfully prepared and contained equal amount of thiol groups and alkyne groups. Mass spectroscopy analysis revealed that the precursor comprised of three components: unreacted dithiols, AB2 monomer and di-ynes (Fig. S2†). HPLC measurements revealed that the precursor contained ∼56% of AB2 monomer, ∼22% of di-thiols and ∼22% of di-ynes. In the 1H NMR spectrum of HPs, the proton signal of CH2 moiety in the propargyl group at 4.7 ppm gradually weakened and the signals at 2.5–2.8 ppm assigned to the protons of CH2SCH2 moiety gradually increased upon reaction time, implying the proceeding of polymerization. The emerging proton signals at 6.1 and 5.6 ppm and the emerging vinyl carbon signals at 119–132 ppm (Fig. S3†) implied the formation of vinyl sulfides generated by monoaddition of thiol to alkyne, indicating the generation of linear units (L).6
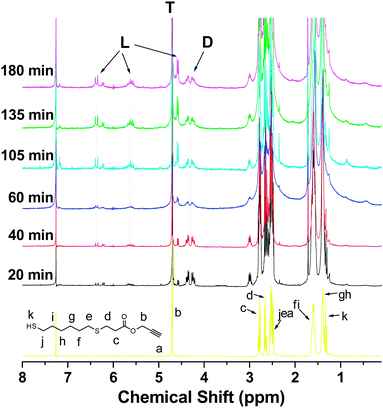 | ||
| Fig. 1
1H NMR spectra of precursor obtained by thiol–ene reaction and corresponding HPs at different polymerization times before precipitation. Here, 6.3, 5.6 and 4.58 ppm, CHCH–CH2OOC moiety of L; 4.7 ppm, CH2OOC moiety of T; 4.2–4.4 ppm, CH2OOC moiety of D. | ||
In order to detect the conversion of thiol groups and probe the structure of the HPs, model compounds were elaborated: Ter was synthesized by thiol–ene addition of 1-propanethiol to propargyl acrylate to mimic the terminal unit (T); Den was prepared by thiol–yne bisaddition of 1-propanethiol to Ter to mimic the dendritic unit (D); two mixtures comprised of Ter, Den and Lin, Mix1 and Mix2 were prepared to mimic the unit structure of HP by inadequate additions of 1.8 and 0.5 equivalent of 1-propanethiol to Ter, respectively (see ESI† for preparation details). Their 1H NMR spectra are shown in Fig. 2. Differences are observed: in the spectrum of Ter, the proton signal of CH2 moiety in the propargyl group is located at 4.7 ppm; in the spectrum of Den, the double peak of the propargyl CH2 moiety originally appearing at 4.7 ppm is converted into the multiple peak at 4.2–4.4 ppm by bisaddition; in the spectrum of Mix1 and Mix2, the vinyl group of Lin formed by monoaddition displays its proton signals at 5.6 and 6.3 ppm, and the adjacent CH2 moiety shows its signal at 4.58 ppm. All these proton signals mentioned here can be seen in the 1H NMR spectra of HPs, justifying that the model compounds can be used to examine the conversion of thiol groups and the unit structure of HP accurately.
 | ||
| Fig. 2 1H NMR spectra of model compounds. Den, model for the dendritic units; Ter, model for the terminal units; Lin, model for the linear units; Mix1 and Mix2, models for the HP. | ||
The conversion of thiol groups and the degree of branching (DB) were calculated according to the following two formulas, respectively.
 | (1) |
 | (2) |
As a result, the conversion of thiol groups reached 95.4% in 3 h, indicating that the polymerization proceeds very fast. All the DB values are higher than 0.5 (the maximum value for a HP made from AB2 monomer without substitution effect) due to that the first addition of thiol to alkyne (r1) is much slower than the second addition of thiol to vinyl sulfide (r2) and thus the reactivity ratio (γ) of r2 to r1 is generally larger than 1.11 In the kinetics theory with substitution effect, if γ ≥ 10, DB will be close to or even higher than 0.7 at conversion of 70%.12
The molecular weights and polydispersity indices (PDIs) of HPs precipitated from methanol were determined by gel permeation chromatography (GPC). HP at 0.33 h of polymerization cannot be precipitated out because of its low molecular weight. GPC curves and polymerization data are presented in Fig. 3 and Table 1, respectively. It can be seen that the elution peaks come out earlier and the curves become broader with increasing the polymerization time. Both Mn (number-average molecular weight) and Mw (weight-average molecular weight) increase rapidly with conversion, with Mw growing much faster than Mn, resulting in higher and higher PDI. This phenomenon could also be seen in the reported preparations of other HPs.2,6Polymerization for 3 h afforded the HP with Mn of 7200, Mw of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4800 and DPw (weight-average degree of polymerization) of 402. It should be pointed out that at the high conversion up to 95.4%, no gelation occurred, implying that the reaction between 22% of di-thiol and 22% of di-yne which has the potential to cause crosslinking at low conversion cannot impose remarkable impact in the presence of 56% of AB2 monomer. This confirms the validity of the A2 + CB2 approach that differs from the conventional AB2 or A2 + B3 approaches in essence.
4800 and DPw (weight-average degree of polymerization) of 402. It should be pointed out that at the high conversion up to 95.4%, no gelation occurred, implying that the reaction between 22% of di-thiol and 22% of di-yne which has the potential to cause crosslinking at low conversion cannot impose remarkable impact in the presence of 56% of AB2 monomer. This confirms the validity of the A2 + CB2 approach that differs from the conventional AB2 or A2 + B3 approaches in essence.
| Sample | Time/min | Conva (%) | M n (k) | M w (k) | PDIb | DB | D | T | L |
|---|---|---|---|---|---|---|---|---|---|
| a Conversion of thiol groups. b Polydispersity index (Mw/Mn). | |||||||||
| HP-A | 20 | 55.4 | — | — | — | 0.98 | 0.27 | 0.71 | 0.02 |
| HP-B | 40 | 67.9 | 2.3 | 3.4 | 1.48 | 0.91 | 0.29 | 0.62 | 0.09 |
| HP-C | 60 | 72.3 | 5.1 | 20.7 | 4.06 | 0.87 | 0.30 | 0.57 | 0.13 |
| HP-D | 105 | 81.5 | 5.3 | 26.3 | 4.96 | 0.82 | 0.32 | 0.50 | 0.18 |
| HP-E | 135 | 92.2 | 6.8 | 87.9 | 12.93 | 0.80 | 0.36 | 0.44 | 0.20 |
| HP-F | 180 | 95.4 | 7.2 | 104.8 | 14.56 | 0.76 | 0.36 | 0.40 | 0.24 |
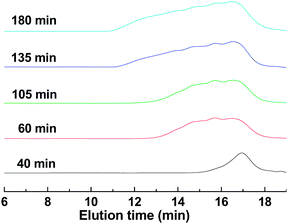 | ||
| Fig. 3 GPC curves of HPs at different polymerization times. | ||
Polymerization under UV irradiation in the presence of 2,2-dimethoxy-2-phenyl-acetophenone (DMPA) was also attempted,6 and yielded HP with Mn of 4500 and Mw of 34300 after irradiation for only 30 min. 3,6-Dioxaoctane-1,8-dithiol was also employed to test the modularity of this SCC procedure, affording HP with Mn of 6300 and Mw of 19300.
Conclusions
In summary, sequential thiol–ene and thiol–yne click chemistry has achieved the rapidness, simplicity and high efficiency during the process from commercial starting materials to final HP product. HPs with high molecular weights and high DBs which both can be adjusted through the reaction time or conversion have been readily prepared from commercial monomers within 12 h in high yield. Both thermo-initiated and UV-initiated thiol–yne polymerizations are available. Currently, employing various functional monomers to create versatile HPs and investigating their applications are in progress in our laboratory. Thiol–halogen click-like and thiol–yne click reactions are successfully combined via the SCC strategy to create HPs with high molecular weights and high DBs, and the results will be published elsewhere. Other click reactions, such as thiol–epoxy, thiol–isocyanate, thiol–ene, azide–alkyne and so on, will also be applied in this sequential procedure, opening an avenue to fast, easy, scalable production of HPs.The prepared hyperbranched polythioether–ynes also have big potential in many applications due to its chemical features, internal thioether groups and peripheral alkyne groups, and dendritic architecture. The sulfur-rich feature benefits the application of the HPs in metal ion absorption, oil resistance, anti-oxidation, and so on. The alkyne groups at the periphery can facilitate the modification of HPs via highly efficient thiol–yne and azide–alkyne click chemistry techniques.
Experimental
The whole preparation procedure for HP can be completed within 12 h. Typically, 1,6-hexanedithiol (1.51 g, 10 mmol) and Et3N (1.01 g, 10 mmol) were dissolved in THF (4 mL) in a 50 mL flask placed in a water bath. The solution was purged with N2 for 10 min to eliminate oxygen and then added dropwise a THF (2 mL) solution of propargyl acrylate (1.10 g, 10 mmol) over 30 min with vigorous stirring. The reaction system was kept at 35 °C and monitored with 1H NMR spectroscopy. It was found that proton signals of the CH2CH moiety of acrylate totally disappeared 6 h later. Then a vacuum pump was connected with the flask to remove THF and Et3N, affording colourless liquid (2.59 g) containing equal amounts of thiol and propargyl groups. 1H NMR (300 MHz, CDCl3): 4.7 (d, 2H, CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2O), 2.82–2.6 (m, 4H, OCCH2CH2S), 2.58–2.49 (m, 4H, HSCH2CH2CH2CH2CH2CH2S), 2.48 (t, 1H, CHC), 1.52–1.68 (m, 4H, HSCH2CH2CH2CH2 CH2CH2S), 1.46–1.35 (m, 4H, HSCH2CH2CH2CH2CH2CH2S), 1.33 (t, 1H, HSCH2). 13C NMR (125MHz, CDCl3): 171.2, 77.8, 75.4, 52.2, 34.8, 33.9, 32.1, 29.5, 28.4, 28.0, 26.9, 24.6. GC-MS: 150.0 g mol−1 for dithiols, 260.1 g mol−1 for AB2 monomer, 371.2 g mol−1 for di-ynes (calculated 150.31 g mol−1, 260.42 g mol−1, and 370.53 g mol−1).
CCH2O), 2.82–2.6 (m, 4H, OCCH2CH2S), 2.58–2.49 (m, 4H, HSCH2CH2CH2CH2CH2CH2S), 2.48 (t, 1H, CHC), 1.52–1.68 (m, 4H, HSCH2CH2CH2CH2 CH2CH2S), 1.46–1.35 (m, 4H, HSCH2CH2CH2CH2CH2CH2S), 1.33 (t, 1H, HSCH2). 13C NMR (125MHz, CDCl3): 171.2, 77.8, 75.4, 52.2, 34.8, 33.9, 32.1, 29.5, 28.4, 28.0, 26.9, 24.6. GC-MS: 150.0 g mol−1 for dithiols, 260.1 g mol−1 for AB2 monomer, 371.2 g mol−1 for di-ynes (calculated 150.31 g mol−1, 260.42 g mol−1, and 370.53 g mol−1).
The resulting liquid was diluted with toluene to 0.5 M and mixed with AIBN (32.8 mg, 0.2 mmol). After purging N2 to the flask for 10 min, the temperature was elevated to 65 °C to start the polymerization. Samples were collected in given reaction time through air-tight syringe and analyzed by 1H NMR spectroscopy. The polymer was precipitated out by pouring the polymer solution into methanol and characterized by GPC. The polymer irradiated for 3 h was characterized by 13C NMR spectroscopy (Fig. S3†).
UV irradiation initiated polymerization was also attempted by employing 2 mol% of DMPA as photoinitiator instead of AIBN.
Acknowledgements
Financial support from NSFC (No. 20974093), the National Basic Research Program of China (2007CB936004), Qianjiang Talent Foundation (2010R10021), the Fundamental Research Funds for the Central Universities (2009QNA4040), Research Fund for the Doctoral Program of Higher Education of China (20100101110049), and Zhejiang Provincial Natural Science Foundation of China (No. R4110175) are gratefully appreciated. Dr J. Han acknowledges China Postdoctoral Science Foundation (No. 20100471707).Notes and references
- P. Flory, J. Am. Chem. Soc., 1952, 74, 2718 CrossRef CAS; B. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef; C. Gao, Hyperbranched Polymers and Functional Nanoscience, Novel Polymers and Nanoscience, ed. M. Adeli, Transworld Research Network, Kerala, India, 2008, ch. 2, p. 33 CrossRef; D. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef; J. Han and C. Gao, Curr. Org. Chem., 2011, 15, 2 CrossRef.
- C. Gao and D. Y. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- C. Gao and D. Y. Yan, Macromolecules, 2003, 36, 613 CrossRef CAS; C. Gao and D. Y. Yan, Macromolecules, 2001, 34, 156 CrossRef; C. Gao and D. Y. Yan, Chem. Commun., 2001, 107 RSC; D. Y. Yan and C. Gao, Macromolecules, 2000, 33, 7693 CrossRef.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751 CrossRef CAS; X. Ma, J. Tang, Y. Shen, M. Fan, H. Tang and M. Radosz, J. Am. Chem. Soc., 2009, 131, 14795 CrossRef; J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Polymer, 2007, 48, 1526 CrossRef; L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, D. E. Drockenmuller, B. M. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063 CrossRef.
- H. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef; R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef; A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522 RSC; A. J. Qin, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2010, 43, 8693 CrossRef; K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276 CrossRef; G. Franc and A. Kakkar, Chem.–Eur. J., 2009, 15, 5630 CrossRef; Z. A. Li, W. B. Wu, Q. Q. Li, G. Yu, L. Xiao, Y. Q. Liu, C. Ye, J. G. Qin and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 2763 Search PubMed; C. Gao and X. Zheng, Soft Matter, 2009, 5, 4788 RSC; J. Y. Wu, H. K. He and C. Gao, Macromolecules, 2010, 43, 2252 CrossRef; J. Y. Wu and C. Gao, Macromolecules, 2010, 43, 7139 CrossRef; Y. Zhang, H. K. He, C. Gao and J. Y. Wu, Langmuir, 2009, 25, 5814 CrossRef.
- D. Konkolewicz, C. K. Poon, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075 CrossRef CAS; D. Konkolewicz, C. K. Poon, A. Gray-Weale and S. Perrier, Chem. Commun., 2011, 47, 239 RSC; W. Liu and C. M. Dong, Macromolecules, 2010, 43, 8447 CrossRef.
- A. J. Scheel, H. Komber and B. I. Voit, Macromol. Rapid Commun., 2004, 25, 1175 CrossRef CAS; Z. A. Li, G. Yu, P. Hu, C. Ye, Y. Q. Liu, J. G. Qin and Z. Li, Macromolecules, 2009, 42, 1589 CrossRef; A. J. Qin, J. W. Y. Lam, C. K. W. Jim, L. Zhang, J. J. Yan, M. Häussler, J. Z. Liu, Y. Q. Dong, D. H. Liang, E. Q. Chen, G. C. Jia and B. Z. Tang, Macromolecules, 2008, 41, 3808 CrossRef; J. D. Xie, L. H. Hu, W. F. Shi, X. X. Deng, Z. Q. Cao and Q. S. Shen, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1140 CrossRef; Z. A. Li, W. B. Wu, G. F. Qiu, G. Yu, Y. Q. Liu, C. Ye, J. G. Qin and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1977 CrossRef.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959 RSC; J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158 CrossRef CAS; B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef.
-
1H NMR spectrum of propargyl acrylate: 5.8–6.5 (CH2CH), 4.76, 2.5.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113 CrossRef CAS.
- Z. P. Zhou and D. Y. Yan, Sci. China Chem., 2010, 53, 2429 CrossRef CAS; Z. P. Zhou and D. Y. Yan, Macromolecules, 2008, 41, 4429 CrossRef; D. Y. Yan, A. H. E. Müller and K. Matyjaszewski, Macromolecules, 1997, 30, 7024 CrossRef; Z. P. Zhou, Z. W. Jia and D. Y. Yan, Polymer, 2009, 50, 5608 CrossRef; Z. P. Zhou and D. Y. Yan, Kinetic Theory of Hyperbranched Polymerization, in Hyperbranched Polymers: Synthesis, Properties, and Applications, ed. D. Y. Yan, C. Gao and H. Frey, Wiley, 2011, ch. 13 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and data. See DOI: 10.1039/c1py00235j |
| This journal is © The Royal Society of Chemistry 2011 |