Synthesis of highly syndiotactic polymers by discrete catalysts or initiators
Garret M.
Miyake†
and
Eugene Y.-X.
Chen
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523-1872, USA. E-mail: eugene.chen@colostate.edu
First published on 3rd August 2011
Abstract
Progress in the synthesis of highly stereoregular polymers with syndiotacticity ≥90% from an array of monomer substrates is surveyed, with a focus being placed on the use of syndiospecific discrete catalyst or initiator systems also exhibiting high activity and efficiency in the polymerization reactions. The monomer scope encompasses nonpolar α-olefins (propylene, styrene, and higher α-olefins), conjugated diolefins, bicyclic olefins, polar conjugated olefins (acrylic monomers such as methacrylates and (meth)acrylamides), and cyclic esters (lactides and β-lactones), while the polymerization catalysts or initiators enabling such syndiospecific polymerizations cover those discrete molecular complexes of main-group, early transition, and lanthanide metals. Several free-radical polymerization systems capable of producing highly syndiotactic polymers are also highlighted.
 Garret M. Miyake | Garret Miyake received his B.S. from Pacific University in 2005 and Ph.D. from Colorado State University in early 2011 under the direction of Professor Eugene Chen. He was an NSF-EAPSI fellow, carrying out his summer research in 2009 in the laboratory of Professor Eiji Yashima at Nagoya University, Japan. Since March 2011, he has been a postdoctoral fellow working with Professor Bob Grubbs at Caltech. His research interest is in the area of polymerization catalysis for the precision synthesis of chiral and sustainable polymers by transition-metal, main-group, and organic catalysts. |
 Eugene Y.-X. Chen | Eugene Chen is a Professor of Chemistry and Engineering (adjunct) at Colorado State University. His current research interests include: precision synthesis of stereoregular polymers through development of new stereoselective catalysts or polymerizations; renewable energy & sustainability through catalytic conversion of nonfood biomass into fuels or chemicals and polymerization of renewable feedstocks into environmentally sustainable polymers; and nanostructured materials through in situ synthesis and synergistic assembly of organic and inorganic nanocomposites or hybrid polymers. |
Introduction
Polymeric materials are the most important synthetic materials to modern society, exhibiting vast ranges of materials properties that enable their application in countless venues. An impressive repertoire of polymerization techniques have been developed for the synthesis of well-defined polymers through various ionic (anionic and cationic),1–4 radical,5–9 and coordination10–13polymerization mechanisms. Such advancements have allowed for the polymerization of diverse classes of monomers to polymers with specific target applications, accessible by the vast range in polymer properties, governed not only by the chemical composition of the monomer building blocks but equally so in how these building blocks are assembled together. This assembly gives rise to the potential to produce stereoregular, crystalline polymers exhibiting enhanced properties, as compared to their non-stereoregular, amorphous counterparts. Thus, the development of rapid and efficient syntheses of target-oriented polymers still represents a major goal in polymer science.This review focuses on the polymer tacticity control by discrete catalyst or initiator systems, a topic of ever growing interest.14 The tacticity of a polymer, or the relative stereoregularity of stereocenters within a polymer's main-chain, determines its overall crystallinity. A tactic polymer folds and packs its chains in an ordered fashion to render crystallinity, while an atactic polymer lacks any long range order and is amorphous. Hence, tacticity is directly related to the polymer's physical and mechanical properties such as melting transition temperature (Tm), glass-transition temperature (Tg), resistance to solvent and fatigue, as well as modulus and impact strength. In the case of water-soluble, stereoregular polyacrylamides, the polymer tacticity also influences the polymer's effectiveness to act as kinetic hydrate inhibitors in oil field applications.15
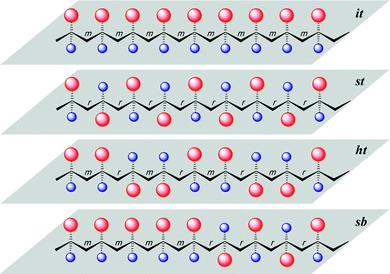
More specifically, this review concentrates on the synthesis of highly syndiotactic polymers by discrete catalyst or initiator systems. Chart 1 depicts mono- and disubstituted tactic vinyl and vinylidene polymers showing the stereogenic center sequence distributions, where mmmmmmmm, rrrrrrrr, mrmrmrmr, and mmmmrrrr correspond to isotactic (it), syndiotactic (st), heterotactic (ht), and it-block(b)-st stereoblock (sb) stereomicrostructures, respectively. Exemplifying the effects that tacticity plays on the properties of a polymer, poly(methyl methacrylate), PMMA, exhibits a wide range of Tg's,16 which are determined by its stereomicrostructure: ca. 55,17 87,17 110,17 130,18 and 140 °C19 for highly isotactic (96% mm), isotactic-b-syndiotactic (46% mm, 46% rr), syndio-biased atactic (60% rr), syndiotactic (81% rr), and highly syndiotactic (95% rr) PMMA, respectively.
This example of greatly enhanced thermal properties of syndiotactic PMMA, as compared to other tactic PMMA, highlights the importance of exploring all stereo-forms of stereoregular polymers, particularly syndiotactic nonpolar and polar polyolefins. The synthesis of highly syndiotactic, technologically important nonpolar polyolefins,20 especially, st-polypropylene (st-PP)21–23 and st-polystyrene (st-PS)24–27 has been thoroughly investigated and reviewed. In 1955, Natta discovered that classical Ziegler–Natta catalysts produced PP containing fractions of highly syndiotactic PP that can be isolated.28 However, highly syndiotactic PS was not achievable by such catalysts, and its first synthesis was realized only 30 years later by Ishihara and co-workers, utilizing half-sandwich organotitanium complexes activated with organoaluminum compounds such as methylaluminoxane (MAO).29–31 Since these initial reports of the synthesis of st-PP and st-PS, numerous successful approaches or catalyst systems have been developed towards their synthesis under industrial conditions and several of them have been commercialized, which have been recently and thoroughly reviewed prior (vide supra). Hence, a comprehensive review on the synthesis of st-PP and st-PS is beyond the scope of this survey, although a brief history and significant recent advancements will be highlighted. In contrast, the synthesis of highly syndiotactic polar conjugated olefin (acrylic) polymers, such as st-PMMA,4 has been limited to low temperature (typically at −78 °C or lower) anionic polymerization, and its synthesis by chiral catalyst-site-controlled coordination polymerization at industrially desirable conditions (i.e. ambient or above temperatures) has been achieved only very recently.10 The ability to synthesize highly syndiotactic polar polyolefins under industrial settings will lead to an expansion of the applications of such polymers.
Accordingly, the goal of this article is to survey reports in the synthesis of highly syndiotactic polymers, where the term “highly syndiotactic” is arbitrarily defined by the authors as a polymer exhibiting racemic triads of rr ≥ 90%. This review is organized by polymer type, including polymers from nonpolar α-olefins (propylene, styrene, and higher α-olefins), conjugated diolefins, bicyclic olefins, polar conjugated olefins (acrylic monomers such as methacrylates and (meth)acrylamides), and cyclic esters (lactides and β-lactones), which is further subdivided, when accomplished, by polymerization mechanism. It should be pointed out that, although most of the catalyst species described herein are only active for polymerization in their cationic or other active forms derived from the activation of the precatalyst with an appropriate cocatalyst (activator), the term catalyst will be used to describe both neutral and cationic or other active forms. In addition, where the term “catalyst” is used for living/controlled coordination polymerization of polar conjugated olefins, it emphasizes the catalyzed monomer enchainment. In this context, it is a catalyst when emphasizing the fundamental catalytic event of monomer enchainment (i.e., the propagation “catalysis” cycle), but it is not a “true” catalyst if the catalytic production of polymer chains is concerned. Furthermore, several other characteristics of the polymerization and polymers will be described, including: polymerization medium (solvent), temperature (Tp), time (t), turnover frequency [TOF, defined as mole of monomer (M) consumed (or polymer formed) per mole catalyst (or initiator, I) per h], and catalyst or initiator efficiency [I*, defined by I* = Mn(calcd)/Mn(exptl), where Mn(calcd) = molecular weight (MW) of M × [M]0/[I]0 × conversion % + MW of end groups and Mn (Mw) = number (weight)-average MW], as well as polymer MW (Mn and Mw) and MW distribution (MWD or polydispersity index, PDI = Mw/Mn). To provide a means of directly comparing different polymerization systems to each other, polymerizationactivity in this review has been converted to TOF values with the same unit of h−1. Lastly, a well-definedpolymer is characterized by a high degree control on its chain structure (regiochemistry and MW characteristics such as Mn, Mw, and MWD), topology (linear, branched, dendritic, etc.), or stereomicrostructure (tacticity or helicity), all of which can have drastic impact on the materials properties of polymer.
1. Polypropylene
Syndiotactic PP is a crystalline thermoplastic material that exhibits lower Tm (150–155 °C) and crystallization rates, but greater impact strength, higher flexibility, and better optical clarity, than its isotactic counterpart, governed not only by the stereoregularity but also through the error sequences which in tandem dictate the crystallization kinetics and morphology.32 In 1988, Ewen and co-workers discovered that Cs-symmetric metallocene dichlorides (1, Chart 2) bearing an isopropylidene (Me2C<) bridged Cp-Flu (Cp = cyclopentadienyl, Flu = fluorenyl) ligand set, upon activation with excess MAO, are highly active catalysts for the syndiospecific polymerization of propylene at ambient or higher temperatures leading to highly syndiotactic, fully regioregular PP.33,34 The polymerization of liquid propylene at 25 °C catalyzed by 1 is highly active (TOF = 4.76 × 105 h−1) and produces st-PP (86% rrrr) with a MW of 133 kDa and a PDI of 1.9. Increasing the polymerization temperature to 50 °C resulted in a dramatic increase in activity by ∼15-fold (TOF = 7.12 × 106 h−1), which was accompanied by a decrease in polymer MW (Mw = 69 kDa) and syndiotacticity (81% rrrr). The activity of the Hf analog at 50 °C is lower than the Zr catalyst, but the resulting polymer MW is more than 11 times higher (Mw = 777 kDa) and the syndiotacticity is noticeably lower (74% rrrr).33 Keeping the same Cs-ligand framework, Razavi revealed that diphenylmethylidene (Ph2C<) bridged catalyst 2 produces st-PP with slightly higher syndiotacticity, but much higher MW (4-fold enhancement), albeit with lower polymerization activity, than the PP produced by 1.35 Bercaw and co-workers examined the propylene polymerization behavior in more detail by this Ph2C< bridged catalyst and found that, at 20 °C, catalyst 2 produced highly syndiotactic PP (97.5% rrrr) with high MW (Mw = 840 kDa, PDI = 2.3) and Tm (147 °C); the activity was also impressive, with a TOF of 1.72 × 106 h−1.36 Increasing the polymerization temperature to 40 °C and 60 °C resulted in variations in polymerization activity (Table 1), but a sharp drop in polymer syndiotacticity to 83.6% rrrr and 81.1% rrrr, and polymer Tm to 137 °C and 132 °C, respectively.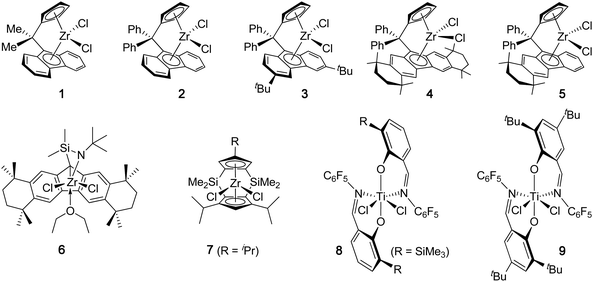 | ||
| Chart 2 Representative discrete catalyst systems that produce highly syndiotactic PP. | ||
| catalyst | T p (°C) | TOF (h−1) | M w (kg mol−1) | PDI (Mw/Mn) | rrrr (%) | T m (°C) | ref. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MAO as cocatalyst for all runs; n.r. = not reported. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 25 | 476,000 | 133 | 1.9 | 86 | n.r. | 33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 50 | 7,121,000 | 69 | 1.8 | 81 | 138 | 33,35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 20 | 1,723,000 | 840 | 2.3 | 97.5 | 147 | 36 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 40 | 265,000 | 380 | 2.0 | 83.6 | 137 | 36 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 60 | 1,192,000 | 270 | 2.0 | 81.1 | 132 | 36 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 40 | n.r. | 766 | n.r. | 91.0 | 150 | 37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 60 | n.r. | 509 | n.r. | 88.5 | 143 | 37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 0 | 105,000 | 961 | 2.1 | >99 (r) | 153 | 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 20 | 259,000 | 843 | 1.8 | >98 (r) | 148 | 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 0 | 13,000 | n.r. | n.r. | >98 (r) | 140 | 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | –15 | 31,000 | 58.6 | 2.25 | >99 | 165 | 39 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 25 | 921,000 | 35.7 | 2.17 | 96 | 157 | 39 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 20 | 4,039,000 | 980 | 2.0 | 97.5 | 151 | 36 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 50 | 17,671,000 | 290 | 2.0 | 81.0 | 123 | 36 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 0 | 72 | 26.7 | 1.08 | 94 (rr) | 156 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 25 | 139 | 50.8 | 1.08 | 93 (rr) | 152 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 50 | 113 | 43.2 | 1.23 | 90 (rr) | 149 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 0 | 245 | 107 | 1.11 | 96 | 148 | 45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Much effort has been directed towards introducing substituents into the Flu ring for further improving the syndiospecicifity of Ph2C< bridged Cs-symmetric metallocene catalysts. In this context, Razavi and co-workers found that catalyst 3 with 3,6-di-tert-butyl substitutions on the Flu ring is indeed more syndiospecific than the parent catalyst 2.37 Thus, the propylene polymerization by 3 at 40 °C yielded st-PP with high MW (Mw = 766 kDa), syndiotacticity (91% rrrr) and Tm (150 °C). Impressively, even at 60 °C, the resulting polymer still had a high syndiotacticity of 88.5% rrrr. Miller and Bercaw developed a series of sterically expanded Flu derivatives of Zr and Hf complexes incorporating an octamethyloctahydrodibenzofluorenyl (Oct) (4) or a tetramethyltetrahydrobenzofluorenyl (5) ligand.38 In toluene at 0 °C, complex 4, when activated with 2000 equiv of MAO, exhibited a good activity of TOF = 1.05 × 105 h−1, producing st-PP with high MW (Mw = 961 kDa, PDI = 2.1), syndiotacticity (r > 99%), and Tm (153 °C). Increasing the polymerization temperature to 20 °C decreased MW (Mw = 843 kDa, PDI = 1.8) and slightly reduced the syndiotacticity and Tm (r > 98%, Tm = 148 °C), but more than doubled the polymerization activity (TOF = 2.59 × 105 h−1). The hafnium analog produced PP with noticeably lower syndiotacticity and with lower activity. The unsymmetrically substituted Flu derivative 5 also produced highly syndiotactic PP at 0 °C (r > 98%) but with lower Tm (140 °C) and activity (TOF = 1.30 × 104 h−1). The Me2C< bridged Oct derivative is less active and syndioselective than the Ph2C< bridged catalyst 4.
The above catalysts 1–5 are characterized as 1,1-disubstituted methylidene (R2C<) bridged Cs-ansa-Cp-Flu metallocene catalysts. Exploring other bridging systems, Irwin and Miller synthesized a sterically expanded, “constrained geometry” type η1-Flu-η1-amidoansa-zirconium complex (6) and found that 6, upon activation with excess MAO, exhibits highly attractive characteristics in the syndiospecific polymerization of propylene.39 At −15 °C, the polymerization of liquid propylene by 6 produced essentially quantitatively syndiotactic PP (rrrr > 99%), which displayed the highest Tm known for st-PP (165 °C for the unannealed polymer, 174 °C for the annealed polymer). The activity of the polymerization at −15 °C was modest (TOF = 3.10 × 104 h−1), producing st-PP also with a modest Mw of 58.6 kDa (PDI = 2.25). Increasing the polymerization temperature to 25 °C resulted in a large increase in polymerization activity by ∼30-fold (TOF = 9.21 × 105 h−1), accompanied by a small drop in the polymer syndiotacticity to rrrr = 96% and Tm to 157 °C and a considerable decrease in MW to Mw = 35.7 kDa. An analogous “constrained geometry” titanium catalyst supported by an η3-3,6-tBu2-Flu-η1-amido ligand is less syndiospecific, affording st-PP with rr = 93% (86% rrrr) at Tp of 0 °C.40 Doubly silylene-bridged Cs-ansa-metallocenes complexes (7), developed by Bercaw and co-workers, are also highly syndiospecific for propylene polymerization.36,41 For example, complex 7 (R = CHMe2), upon activation with 2000 equiv of MAO in toluene at 20 °C, was highly active and syndiospecific for propylene polymerization (TOF exceeding 4.0 × 106 h−1), producing st-PP with rrrr = 97.5% and Tm = 151 °C. Increasing the polymerization temperature to 50 °C led to a drastic increase in polymerization activity, with TOF exceeding 1.7 × 107 h−1, at the expense of the much reduced syndiotacticity (81% rrrr) and Tm (123 °C). Complexes with other substitutents (e.g., R = H, SiMe3) also produced highly syndiotactic PP at 20 °C with rrrr ∼ 94% and Tm ∼ 151 °C.
Departing from the prototype Cs-symmetric ansa-metallocene catalyst system, where the propylene polymerization by Zr catalysts is much more effective and syndiospecific than analogous Ti and Hf catalysts and proceeds via 1,2-insertion regiochemistry and catalyst-site-control stereochemistry, Fujita and co-workers have developed a class of C2-symmetric, octahedral non-metallocenetitanium catalysts supported by phenoxy-imine chelating ligands (FI catalysts) that polymerize propylene in a highly syndiospecific and living fashion via 2,1-insertion regiochemistry and chain-end-control stereochemistry.42 Thus, catalyst 8 with a bulky trimethylsilyl grouportho to the phenoxy-O and a C6F5 group on the imine-N produced highly syndiotactic PP with rr = 94%, 93%, 90% and Tm = 156 °C, 152 °C, 149 °C, for the polymerizations activated with MAO and carried out at 0 °C, 25 °C, and 50 °C, respectively.43 However, the activity of <140 h−1 is lower than the prototype zirconocene catalysts by several orders of magnitude (cf.Table 1). Replacing the orthoTMS group in 8 by the tBu group experienced considerable reductions in all aspects of the polymerization at 25 °C, including activity (TOF = 87 h−1), syndiotacticity (rr = 87%), melting transition (Tm = 137 °C), and MW (Mw = 31.6 kDa). In fact, the syndiotacticity of the PP produced at 25 °C by the derivatives of catalyst 8 with different R subsitituents drops linearly on going from TMS (93% rr), tBu (87% rr), iPr (75% rr), Me (50% rr), and H (43% rr). Coates and co-workers have developed analogous phenoxy-imine titanium catalysts for the synthesis of st-PP.44 With an additional tBu group placed on the phenoxy-benzene ring, now titanium catalyst 9 (activated by MAO) with the tBu grouportho to the phenoxy-O can also produce highly syndiotactic PP at 0 °C, with rrrr = 96%, Mw = 107 kDa, PDI = 1.11, Tm = 148 °C, and enhanced activity (TOF = 245 h−1).45
Unlike the above C2-symmetric bis(phenoxy-imine) titanium catalysts that polymerize propylene syndiospecifically via 2,1-insertion regiochemistry and chain-end-control stereochemistry, Cs-symmetric, R2C< bridged ansa-Cp-Flu metallocene catalysts polymerize propylene syndiospecifically via 1,2-insertion regiochemistry and catalyst-site-control stereochemistry. Chart 3 depicts the proposed propagating species and stereocontrol that involves the alternating, migratory insertion of propylene at the enantiotopic sites of the Cs-ligated cationic metal complex. Several pathways can introduce stereoerrors, including enantiofacial misinsertion (mm type) as the predominant source of stereodefects, site epimerization (m type), “back-side” misinsertion (mr type), and chain epimerization (m or mm type).46 Hence, the resulting polymer syndiotacticity is sensitive to polymerization conditions (e.g., solvent polarity, temperature, monomer concentration) and ion-pairing characteristics (e.g., cation and anion structure, coordination, and dynamics).
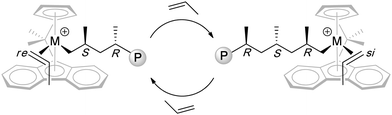 | ||
| Chart 3 Proposed propagating species and catalyst-site stereocontrol in the syndiospecific polymerization of propylene by Cs-ligated catalysts. | ||
2. Polystyrene
In contrast to PP, syndiotactic PS has a higher rate of crystallization than that of other tactic PS configurations and also a considerably higher Tm (by ∼40 °C) than that of it-PS.47 Thus, st-PS, while maintaining a near constant Tg of 100 °C, exhibits a Tm ranging from 265 °C to 275 °C, depending on the polymorphic phase(s) in presence; indirectly, this is dependent on catalyst structure and polymerization conditions. Highly syndiotactic PS is also much more resistant to common solvents,48 and the insolubility of st-PS in boiling ketones is often used as a technique to demonstrate that the polymer is highly syndiotactic (rrrr > 98%).26 The combination of heat and chemical resistance with good electrical properties (low dielectric constant and dissipation factor), coupled with a low polymer density, makes st-PS an attractive material for applications in automotive, electrical and electronics, as well as industrial and consumer uses.49,50 Although st-PS was first synthesized in 1985,29 these attractive polymer properties have upheld strong motivation in developing new catalysts or approaches towards its synthesis, and thus the field has grown rapidly since that time, with half-titanocene complexes activated with MAO achieving the majority of the success.In their seminal report, Ishihara and co-workers described the synthesis of st-PS through the use of CpTiCl3 and a large excess of MAO as cocatalyst or activator.30,31 The polymerization in toluene at 50 °C proceeded to a quantitative monomer conversion, but the activity decreased during the course of polymerization (20 min, TOF = 3.37 × 104 h−1; 180 min, TOF = 1.34 × 104 h−1), indicative of catalyst deactivation. The PS produced is highly syndiotactic (98 wt% insoluble in boiling methyl ethyl ketone) and has a high Tm of 266 °C and a Mw of 63.3 kDa (PDI = 1.99), Table 2; the Mn of the polymer remained nearly constant at different monomer conversions, due to chain transfer reactions present in this polymerization. Among a number of metal complexes screened, only titanium-based complexes can produce st-PS. Under these conditions, homoleptic non-titanocene compounds gave low monomer conversions (<10%), but half-titanocene(III), CpTiCl2, achieved a much higher conversion of 44%, while half-titanocenes(IV), CpTiCl3 (10) and Cp*TiX3 (11, X = Cl, Cp* = pentamethyl cyclopentadienyl, Chart 4), enabled quantitative monomer conversion and produced highly syndiotactic PS.31 Sandwich titanocenes such as Cp2TiCl2 and Cp*2TiCl2 offered only very low yield (≤2%) of st-PS.
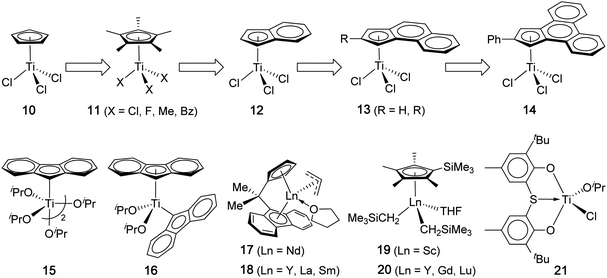 | ||
| Chart 4 Representative discrete catalyst systems that produce highly syndiotactic PS. | ||
| Catalyst | Cocatalyst (equiv) | T p (°C) | TOF (h−1) | M w (kg mol−1) | PDI (Mw/Mn) | SY %a (rrrr %) | T m (°C) | ref. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Syndiotactic yield, measured by wt% insoluble st-PS (rrrr > 98%) in boiling ketones such as 2-butanone. The r pentads in the parenthesis were determined by 13C NMR. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | MAO (600) | 50 | 33,700 | 63.3 | 1.99 | (98) | 266 | 30,31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11-Cl | MAO (300) | 50 | 140 | 169 | 3.6 | n.r. | 275 | 51 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11-F | MAO (300) | 50 | 6,640 | 660 | 2.0 | n.r. | 275 | 51 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11-Me | B(C6F5)3 | 25 | 2,080 | 268 | 2.6 | n.r. | n.r. | 53 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11-Bz | B(C6F5)3 | 25 | 900 | 122 | 1.7 | n.r. | n.r. | 53 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11-Me | Ph3CB(C6F5)4 | 50 | 3,430 | n.r. | n.r. | 96 | n.r. | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | MAO (4000) | 50 | 15,690 | 720 | n.r. | 98.2 | 271 | 56,57 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13-H | MAO (4000) | 50 | 71,920 | 545 | n.r. | 92.5 | 270 | 58 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13-Me | MAO (4000) | 50 | 76,150 | 424 | n.r. | 92.8 | 275 | 58 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | MAO (4000) | 75 | 317,310 | 130 | n.r. | 92 | 265 | 59 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | MAO (500) | 50 | 11,730 | 324 | 1.90 | >99 | n.r. | 62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | MAO (500) | 50 | 29,520 | 400 | 2.17 | >99 | n.r. | 62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | none | 60 | 16,440 | 93.4 | 1.73 | (>99) | 264 | 63 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | Ph3CB(C6F5)4 | 25 | 130,940 | 519 | 1.37 | (>99) | 273 | 66 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21 | MAO (150) | 80 | 17,120 | 269 | 4.8 | >95 | 268 | 70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Upon activation with excess MAO, half-titanocene complexes of a general formula CpTiX3 (X = halides, alkoxides, alkyls, etc.) are highly active, similarly syndiospecific styrene polymerization catalysts, but the X ligand can significantly impact the polymerization activity and the resulting polymer MW, with fluorides being typically the most active, followed by alkoxides and then chlorides.26 For instance, the polymerization activity of Cp*TiF3 (11, X = F) is enhanced by 46-fold over Cp*TiCl3 (11, X = Cl) under the same conditions, with both producing highly syndiotactic PS having a Tm of 275 °C (Table 2).51 In addition, the MW (Mw = 660 kDa, PDI = 2.0) of the st-PS produced by Cp*TiF3 is ∼ four times higher than the MW of the polymer produced by the chloride analog. The observed significant role of the X ligands in the styrene polymerization activity and the polymer MW is presumably attributed to the relative abstractive kinetics (activation by MAO), stability of the resulting cationic catalyst (which is influenced by the coordinating nature of the counterion [X–MAO]−), and propensity towards β-H elimination. Alkyl derivatives such as Cp*TiMe3 and Cp*TiBz3 need only a stoichiometric amount of molecular cocatalysts such as B(C6F5)3 and [Ph3C][B(C6F5)4] to generate the corresponding syndiospecific cationic catalysts (Table 2).52–55 The structural characterizability of the cationic catalyst resulted from the above abstractive activation using a stoichiometric amount of the molecular cocatalyst has also provided important information about the mechanistic aspects of the polymerization.
Extending the supporting ligand from Cp to Ind (Ind = η5-indenyl), Chien, Rausch and co-workers observed the enhanced polymerization activity (by 50–100%) and higher syndiotactic yield of the bulk PS when comparing (Ind)TiCl3 (12) with CpTiCl3 (10) under identical polymerization conditions (including the amount of MAO, temperature, time, and concentrations of the monomer and catalyst).56,57 The authors attributed the observed higher activity and syndiotactic yield (up to 98.2%) by (Ind)TiCl3 to the greater electron-donating ability of the indenyl ring relative to the Cp ring. The more thermally robust catalyst supported by Ind also enabled its polymerization at high temperature (75 °C) without noticeable loss in activity and syndiotactic yield. Since this finding, a large number of half-titanocene catalysts supported by substituted indenyl derivatives have been developed,26 with catalysts 1358 and 1459 standing out for their significantly improved activity (Table 2). As in the case of Cp*-based complexes, (Ind)TiF3 shows significant enhancements in polymerization activity as well as the resulting polymer MW and Tm, relative to (Ind)TiCl3.60 Naturally extending the supporting ligand to η5-fluorenyl (Flu) is of interest, but (Flu)TiCl3 is thermally unstable.61 Knjazhanski and co-workers solved this issue by preparing stable Flu-based mono- and bis-Flu titanium isopropoxide complexes [(η1-Flu)Ti(OiPr)2(μ-OiPr)]2 (15) and (η1-Flu)(η5-Flu)Ti(OiPr)2 (16).62 Both catalysts are highly active, with the η5-Flu-ligated catalyst 16 being 2.5 times more active than the η1-Flu-ligated catalyst 15, reaching a TOF of 2.95 × 104 h−1 (Table 2). Both Flu-based catalysts produce st-PS with a near quantitative syndiotactic yield of >99%, slightly outperforming the Cp and Ind isopropoxide derivatives, which produced PS with a syndiotactic yield of 95% and 98%, respectively, in a comparative study carried out under the same conditions.
Departing from the intensively investigated half-titanocene-type catalysts, in 2004 Carpentier and co-workers synthesized ansa-lanthanocene allyl complexes (17, Ln = Nd; 18, Ln = Y, La, Sm) incorporating the Cs-symmetric Me2C(Cp)(Flu) ligand set, which had been proven effective in the syndiospecific polymerization of propylene, and probed their effectiveness as single-component (i.e., without cocatalysts) syndiospecific styrene polymerization catalysts.63 The styrene polymerization was conducted under neat conditions or in toluene at 60 °C, with all lanthanide catalysts producing highly syndiotactic PS (rrrr ≥ 99% by 13C NMR). The polymerization activity follows the order Nd ≫ Sm > La > Y, with the most active Nd catalyst (17) reaching a TOF of 1.64 × 104 h−1 (Table 2). Soluble syndiotactic oligostyrenes were prepared recently using the Nd catalyst in combination with nBu2Mg that promotes a coordinative chain transfer polymerization, thus producing up to 130 PS chains per Nd.64 The syndiospecific polymerization by 17 was shown proceed predominantly by a chain-end control mechanism, and the copolymerization of styrene and ethylene by the Nd catalyst afforded st-PS-co-PE copolymers composed of st-PS segments randomly separated by isolated ethylene units.65 On the other hand, Hou and co-workers found that neutral half-sandwich lanthanocenes, (C5Me4SiMe3)Ln(CH2SiMe3)2(THF) (19, Ln = Sc; 20, Ln = Y, Gd, Lu), are inactive for styrene polymerization at room temperature in toluene; however, upon activation with 1 equiv of Ph3CB(C6F5)4, all resulting cationic complexes are active for the polymerization, producing quantitatively syndiotactic PS with Tm = 268–273 °C.66 Within this series, the cationic Sc catalyst derived from the precursor 19 exhibits the highest activity (TOF of 1.31 × 105 h−1 at 25 °C), which is comparable with the most active half-titanocene catalysts and higher than the other lanthanide-based catalysts derived from 20 in the series by about three orders of magnitude. This half-sandwich cationic Sc catalyst also exhibits some “living” polymerization characteristics, as evidenced by a nearly linear increase in polymer MW as an increase in the monomer-to-catalyst ratio while the polymer MWD remaining narrow. Uniquely, catalyst system 19 also rapidly and selectively copolymerizes styrene and ethylene to produce blocky st-PS-b-PE copolymers that contain large amounts (up to 87 mol%) of racemically enchained styrene sequences (blocks) connected by repeated ethylene units incorporated only in small amounts.66 Recent variations of Cp*-type half-sandwich Sc catalyst precursors included bis(borohydrido)67 and bis(silylamido)68 Sc complexes which, upon activation with Al(iBu)3/Ph3CB(C6F5)4, also produce highly syndiotactic PS. Turning to non-metallocene catalysts, chelating bis(phenolate) titanium complexes, upon activation with MAO, also exhibit good activity for the synthesis of highly syndiotactic PS.69 For instance, titanium complex 21, activated with only 150 equiv of MAO, polymerized styrene at 80 °C with a TOF of 1.71 × 104 h−1, producing PS with a syndiotactic yield of >95% and a Tm of 268 °C.70
Investigations into the nature of active species derived from the half-titanocene + activator (MAO, B(C6F5)3, [HNMe2Ph][B(C6F5)4]) system by ESR,71–73NMR,74 as well as structural model and polymerization75 studies have led to a general consensus that the active species responsible for the formation of st-PS is a cationic TiIII, represented by a general structure of [Cp'TiR]+. Although reactions involving Ti/Al ligand exchange, Ti ligand abstraction by the Lewis acid, and Ti(IV) reduction to Ti(III) can be envisioned to participate in the catalyst formation, the fundament reaction steps leading to the catalyst are still unclear. As the stereomicrostructure of the st-PS produced by CpTiCl3/MAO consisted of long sequences of racemic dyads with only isolated meso dyads, …rrrrrrrmrrrr…, the polymerization is described to proceed via a chain-end control mechanism. A key feature of this chain-end stereocontrol is the steric repulsion between the phenyl groups of the coordinated monomer and the last inserted monomer, which leads to the syndiotactic configuration.26 More specifically, a syndiospecific insertion step involves η2-styrene coordination, ηn (n ≥ 3) benzyl coordination of the last inserted monomer unit of the growing chain, cis-addition to the double bond, secondary (2,1-) styrene insertion, and inversion of the chirality at Ti after each insertion step (Chart 5).76,77 Stereocontrol of the Cs-symmetric ansa-lanthanocene catalyst system 17 was recently investigated by DFT calculations, which showed that the control by the thermodynamics is a result of combined steric repulsions between styrene–styrene and the last-inserted styrene (phenyl ring)–the fluorenyl ring.78
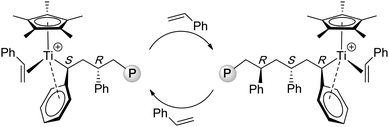 | ||
| Chart 5 Proposed propagating species and chain-end stereocontrol in the syndiospecific polymerization of styrene by half-titanocene catalysts. | ||
3. Poly(methacrylate)s
3.1 Radical polymerization
Successful examples utilizing radical polymerization techniques to produce highly syndiotactic methacrylic polymers are scarce and usually require low to extremely low temperatures to overcome the poor stereochemical control generally associated with radical polymerization mechanisms. In 1970, Farmer and co-workers reported the syndiospecific radical polymerization of methacrylic acid (MAA), initiated by cobalt 60 γ radiation.79 The PMAA samples were esterfied by reaction with diazomethane to afford PMMA for NMR analysis. Investigating solvent interactions and effects of the solvent structure, the authors showed that hydrogen bonding between alcohol solvents and the acidic monomer and the polymer radical provide large steric effects that are amplified at low temperatures, effectively dictating the stereoregularity of the resulting polymer. Specifically, at −78 °C, the polymerization in methanol, 1-propanol, and 2-propanol resulted in polymers with rr = 83.8, 87.1, and 95.0%, respectively. Lowering Tp to −115 °C in 1-propanol increased the syndiotacticity to rr = 92.0%. Chovino and Gramain synthesized poly(methacrylic tetraethyl ammonium salt) using the radical polymerization initiated by 4,4′-azobis(4-cyanovaleric acid) in water.80 Impressively, at a relatively high temperature of 5 °C, highly syndiotactic (rr = 92.0%) polymer was synthesized, which the authors attributed to repulsion between the negatively charged carboxylate groups of the incoming monomer and growing polymer chain, leading to racemic monomer insertion. Okamoto and co-workers utilized fluoroalcohols to influence the stereospecificitiy in the radical polymerization of MMA at low temperatures, initiated by nBu3B/air.81 Thus, the MMA polymerization in (CF3)3COH solvent at −78 °C produced highly syndiotactic PMMA (rr = 91%) in quantitative yield, with Mn = 117 kDa and a relatively high PDI of ∼4. Lowering the reaction temperature to −98 °C only slightly increased the syndiotacticity (rr = 93%), but drastically reduced the polymer yield to only 27%, affording a polymer with Mn = 227 kDa and a very high PDI of ∼9. The authors also showed that addition of even a small amount of (CF3)3COH (0.1 to 1 equiv) to the bulk MMA polymerization was as effective in controlling tacticity as using (CF3)3COH as solvent, and demonstrated that this control over syndiotacticity was largely steric based, due to the H-bonding interaction of the fluoroalcohol with the carbonyl oxygen of the monomer and the propagating species. More recently, Okamoto, Kakuchi and co-workers reported atom-transfer radical polymerization of MMA in fluoroalcohol solvent ((CF3)2CHOH) at low temperatures (–20 °C to −78 °C), affording st-PMMA (rr up to 84%) with Mn ∼ 13 kDa and PDI = 1.17–1.31.82The syndiospecific radical polymerization at elevated temperatures is difficult, but Akashi et al. developed a novel template strategy to synthesize highly isotactic and syndiotactic PMMA, at high temperatures, utilizing the affinity for isotactic and syndiotactic PMMA to form a stereocomplex (Chart 6).83 In their report, the authors prepared silica particles that were coated with porous thin films of either highly isotactic or syndiotactic PMMA, and performed the stereospecific radical polymerization in the presence of these templates; subsequent selective solvent extraction can separate the desired tactic polymer. To synthesize highly syndiotactic PMMA, the authors first polymerized MAA, initiated by 2,2′-azobis(N,N-dimethyleneisobutyramidine)dichloride in water at 40 °C in the presence of silica particles coated with isotactic PMMA, followed by methylation to afford syndiotactic PMMA with rr as high as 98%, but with a rather high PDI of ∼2. Another example of the syndiospecific radical polymerization at elevated temperatures is the radical polymerization of 9-fluorenyl methacrylate, initiated by AIBN. The polymerization was carried out at 60 °C, producing a high MW polymer (Mn = 123.8 kDa, PDI = 1.98) with syndiotacticity rr = 89%.84
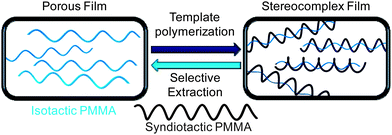 | ||
| Chart 6 Synthesis of highly syndiotactic PMMA by stereocomplex template approach. | ||
3.2 Anionic polymerization
The stereospecificity in the typical anionic polymerization of (meth)acrylates is extremely sensitive to variables such as Tp and solvent, as demonstrated in an early report by Evans and co-workers who investigated the effects of solvent on the anionic polymerization of MMA initiated by fluorenyllithium.85 In mixed toluene-diethyl ether solvent, the polymerization produces isotactic polymers; in mixed toluene-tetrahydrofuran solvent, at low tetrahydrofuran ratios isotactic polymer is formed, whereas when higher tetrahydrofuran ratios (15 vol %) are employed, syndiotactic PMMA is formed. Methacrylates bearing bulky side groups, such as 9-fluorenyl methacrylate, follow this pattern: the polymerization initiated by tBuLi at −78 °C gave an isotactic polymer (m = 90%) in toluene, but a syndiotactic polymer (rr = 89%) in THF.84 Overall, the sensitivity of the resulting polymer tacticity to temperature is due to the chain-end control nature of the typical anionic polymerization, while the solvent effect is attributed to competition between counterion coordination to chain-end (penultimate ester group) and monomer vs. solvation.86 Thus, polar coordinating solvents (e.g., THF, DME) strongly solvate the counterion prohibiting it from exerting an influence on monomer enchainment because it is largely unassociated with the propagating enolate chain end when approaching the monomer, thereby favoring syndiotactic placement for steric reasons, whereas nonpolar solvents (e.g., toluene) typically favor isotactic placement through a rigid propagating chain model involving monomer pre-coordination to the counterion which is associated with the propagating chain end and additionally coordinated to the penultimate ester group.Major efforts made to the anionic polymerization initiated by classical initiators such as organolithium reagents have been to render the polymerization controlled or living by shutting down or significantly suppressing side and/or termination reactions.2,3,87–89 One strategy, developed by Kitayama, Hatada and co-workers, combines alkyl lithium reagents with bulky organoaluminum compounds, which in particular has proven extremely versatile and attractive due to its high degree of control over both polymerization and stereochemistry.90,91 For instance, the MMA polymerization by tBuLi at −78 °C in toluene yielded it-PMMA (mm = 78%) with a high PDI of 3.10 as expected, but highly syndiotactic PMMA (rr = 92%) with a low PDI of 1.13 when combining tBuLi with 3 equiv of trioctylalumunium [Al(nOct)3], Table 3. Decreasing Tp to −93 °C, coupled with 5 equiv of Al(nOct)3, resulted in a noticeable increase in syndiotacticity to rr = 96% (PDI = 1.18).90 The polymerization activity was rather low with TOF ≤ 2 h−1, and the polymerization of other alkyl methacrylates (R = ethyl, isopropyl, and n/ibutyl) by the tBuLi + Al(nOct)3 system also produces highly syndiotactic polymers with narrow MWD's,91Table 3. Aluminum phenoxides, RAl(2,6-tBu2C6H3O)2 (22, Chart 7), can also profoundly affect the stereochemical control of the polymerization by tBuLi at low temperatures. Thus, the polymerization of MMA by tBuLi/22a (5 equiv) at −78 °C afforded ht-PMMA (67.8% mr)92 when R = Me, or st-PMMA (89.1% rr) by tBuLi/22b (5 equiv) when R = Et.93 This strategy was also applicable to the syndiospecific polymerization of trimethylsilyl methacrylate by tBuLi at −78 °C in toluene, yielding a highly syndiotactic polymer with rr = 93.4% or 96.4% when 3 or 5 equiv of 22a was combined with the initiator.94
| Initiator/catalyst | R, [M]/[I] | T p (°C) | TOF (h−1) | M n (kg mol−1) | PDI (Mw/Mn) | rr (%) | T g (°C) | ref. |
|---|---|---|---|---|---|---|---|---|
| t BuLi/3Al(nOct)3 | Me, 50 | –78 | 1.9 | 4.87 | 1.13 | 92 | n.r. | 90 |
| t BuLi/3Al(nOct)3 | i Bu, 50 | –78 | 1.9 | 8.69 | 1.06 | 93 | n.r. | 90 |
| t BuLi/5Al(nOct)3 | Me, 50 | –93 | 1.6 | 4.92 | 1.18 | 96 | n.r. | 90 |
| t BuLi/5 22b | Me, 50 | –78 | 1.7 | 4.45 | 1.10 | 89 | n.r. | 93 |
| t BuLi/5 22b | Et, 50 | –78 | 2.0 | 6.49 | 1.09 | 92 | n.r. | 93 |
| t BuLi/5 22a | TMS, 50 | –78 | 2.0 | 7.03 | 1.16 | 96 | n.r. | 94 |
| 23a/2Al(C6F5)3 | Me, 200 | –78 | 53 | 37.0 | 1.35 | 94 | 138 | 19 |
| t BuLi/2Al(C6F5)3 | Me, 200 | –78 | 42 | 59.8 | 1.33 | 95 | 140 | 98 |
| MTS/0.05Tf2NH | Me, 100 | –55 | <1 | 14.0 | 1.04 | 90 | n.r. | 99 |
| Ph3P/AlEt3 | Me, 40 | –93 | ∼1 | 19.4 | 2.03 | 95 | 135 | 100 |
| 24 | Me, 46.7 | –78 | 521 | 300 (Mw) | n.r. | 93 | n.r. | 102 |
| 25 | Me, 46.7 | –78 | 532 | 120 (Mw) | n.r. | 93 | n.r. | 102 |
| 26 | Me, 10 | –98 | <1 | 17.3 | 1.20 | 94 | n.r. | 103 |
| 26 | Me, 10 | –110 | <1 | 14.0 | 1.19 | 97 | n.r. | 103 |
| 27 | Me, 400 | –30 | 2,280 | ∼40 | ∼1.1 | 92 | 135 | 104 |
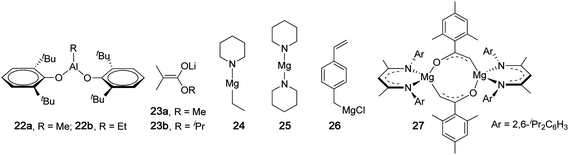 | ||
| Chart 7 Structures of catalysts or anionic initiators that produce highly syndiotactic PMMA. | ||
Lithium ester enolates, Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OR)OLi (alkyl α-lithioisobutyrate, 23), should be, in principle, ideal initiators for the polymerization of (meth)acrylates. As the propagating species in the anionic polymerization of (meth)acrylates by organolithium initiators are lithium ester enolates, rates of initiation and propagation should be nearly identical, giving rise to polymers with narrow MWD's. In actuality, lithium ester enolates have a strong tendency for aggregation (n = 2–6) in hydrocarbon solvents, and the existence of various aggregated ester enolates creates significant problems in controlling the polymerization rate and the polymer MWD.95 Additionally, lithium ester enolates are unstable, even in the solid state, and subject to decomposition to ketenes and lithium alkoxides and β-keto ester enolates.96 Addressing these obstacles, Rodriguez and Chen extended the concept of adding bulky aluminum Lewis acids to organolithium initiators towards the polymerization of methacrylates initiated by lithium enolates and found that the added bulky aluminum species, such as MeAl(BHT)2 (BHT = butylated hydroxytoluene = 4-Me-2,6-tBu2C6H2O), serve as both the catalyst for monomer activation and deaggregator for converting the oligomeric, multi-site enolate species to a monomeric enolaluminate active species.97 The end result is the termed “single-site anionic polymerization” that propagates in a controlled, bimetallic fashion, Chart 10, producing st-PMMA (71% rr) with narrow MWD (1.12) at 23 °C. It should be noted that lithium ester enolaluminates are less reactive than the parent lithium ester enolates, but more selective with preferential addition to the activated monomer in a syndioselective fashion (Chart 8).
C(OR)OLi (alkyl α-lithioisobutyrate, 23), should be, in principle, ideal initiators for the polymerization of (meth)acrylates. As the propagating species in the anionic polymerization of (meth)acrylates by organolithium initiators are lithium ester enolates, rates of initiation and propagation should be nearly identical, giving rise to polymers with narrow MWD's. In actuality, lithium ester enolates have a strong tendency for aggregation (n = 2–6) in hydrocarbon solvents, and the existence of various aggregated ester enolates creates significant problems in controlling the polymerization rate and the polymer MWD.95 Additionally, lithium ester enolates are unstable, even in the solid state, and subject to decomposition to ketenes and lithium alkoxides and β-keto ester enolates.96 Addressing these obstacles, Rodriguez and Chen extended the concept of adding bulky aluminum Lewis acids to organolithium initiators towards the polymerization of methacrylates initiated by lithium enolates and found that the added bulky aluminum species, such as MeAl(BHT)2 (BHT = butylated hydroxytoluene = 4-Me-2,6-tBu2C6H2O), serve as both the catalyst for monomer activation and deaggregator for converting the oligomeric, multi-site enolate species to a monomeric enolaluminate active species.97 The end result is the termed “single-site anionic polymerization” that propagates in a controlled, bimetallic fashion, Chart 10, producing st-PMMA (71% rr) with narrow MWD (1.12) at 23 °C. It should be noted that lithium ester enolaluminates are less reactive than the parent lithium ester enolates, but more selective with preferential addition to the activated monomer in a syndioselective fashion (Chart 8).
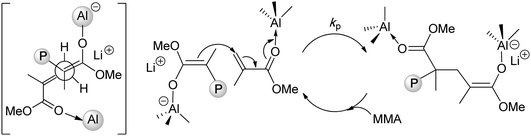 | ||
| Chart 8 Single-site, syndioselective anionic polymerizationvia bimolecular propagation regulated by aluminum Lewis acids. | ||
Among a series of bulky aluminum Lewis acid catalysts examined, the alane Al(C6F5)3 not only generates the most active system (having the highest degree of activation towards monomer), but also renders the system the highest degree of control over polymerization initiated by Me2CC(OiPr)OLi (23b) at ambient temperature or 0 °C.97 At −78 °C, the MMA polymerization by a combination of 23a with 2 equiv of Al(C6F5)3 produced highly syndiotactic PMMA with rr = 94% and Tg = 138 °C.19 Similarly, a combination of tBuLi with 2 equiv of Al(C6F5)3 also afforded highly syndiotactic PMMA (95% rr) at Tp of −78 °C (Table 3). The use of more stable alkyl α-lithioisobutyrates such as 23b, coupled with Al(C6F5)3, affords a more controlled polymerization system than 23a/2Al(C6F5)3, and addition sequence is important for the polymerization by tBuLi + 2 Al(C6F5)3 as premixing these two reagents before addition of MMA generates a hydride-bridged bis(aluminate) initiator, Li[Al(C6F5)3–H–Al(C6F5)3]−, which also affords highly syndiotactic PMMA (94% rr) at Tp of −78 °C.98 Kakuchi et al. recently employed a Brønsted acid, trifluoromethanesulfonimide (Tf2NH), as activator for the anionic polymerization of MMA initiated by 1-methoxy-1-trimethylsilyloxy-2-methylpropene (MTS), which produced st-PMMA (rr = 90%) at −55 °C in DCM.99 This polymerization has a high degree of control, but its activity is very low (TOF < 1 h−1); increasing Tp to 27 °C drastically increased the rate of polymerization (TOF = 10 h−1), at the expense of syndiotacticity (rr = 72%).
In 1992, Hatada and co-workers reported using tertiary phosphines coupled with triethylaluminum for the syndiospecific polymerization of MMA in toluene at low temperatures (–78 to −93 °C).100 Most notably, the Ph3P-Et3Al system polymerized 40 equiv of MMA at −93 °C to st-PMMA (Mn = 19.4 kDa, PDI = 2.03, and rr = 95%) in 70% yield after 24 h. With a needed high 2.5 mol % catalyst loading, the polymerization was sluggish (TOF ∼ 1 h−1) and inefficient (I* < 15%). Performing the polymerization at −78 °C decreased syndiotacticity to 89% rr and diminished the initiator efficiency (I* ∼5%). Utilizing Et3P at −93 °C or −78 °C afforded PMMA with rr = 89% or 81%, with significantly enhanced initiator efficiencies. This system was also shown to polymerize other methacrylates to syndiotactic (∼90% rr) homo- and copolymers.
There has been growing interest in utilizing organo magnesium and calcium compounds in catalysis,101 and in some cases, such species have found success in producing highly syndiotactic PMMA. An early report by Joh and Kotake disclosed that Mg initiators incorporating a piperidine moiety, such as ethylpentamethyleneiminomagnesium (24) and bis(pentamethylenimino)magnesium (25) produced st-PMMA with rr = 93% at −78 °C in toluene.102 Hatada et al. reported a Grignard reagent, m-vinylbenzylmagnesium chloride (26), polymerized MMA in THF from −78 °C to −110 °C, affording highly syndiotactic polymers with rr = 90.6–96.6% with relatively narrow MWD's from 1.30 to 1.19 for the methanol-insoluble fraction (35–75 wt%, depending on Tp).103 More recently, Gibson and co-workers synthesized a discrete magnesium ketone enolate dimer, [(BDI)Mg(μ-OC(CH2)-2,4,6-Me3C6H2)]2 [BDI = HC(C(Me)N-2,6-iPr2C6H3)2] (27, Chart 7), and found it to be highly active for MMA polymerization (TOF = 2,280 h−1) in a living fashion at −30 °C in toluene or chloroform, leading to highly syndiotactic PMMA (92% rr) with a high Tg of 135 °C (Table 3).104 As addition of donor ligands such as THF readily breaks the dimer to form the monomeric Mg enolate complex, it can be envisioned that the MMA polymerization by the dimeric enolate complex 27 proceeds via the monomeric enolate complex in the presence of MMA. The stereocontrol ability of this catalyst system relies on the steric bulk of the ligand rendered by the N-aryl groups; thus, changing from 2,6-iPr2C6H3 to 2,6-Et2C6H3 and 2,6-Me2C6H3N-aryl groups resulted in a substantial reduction of the syndiotacticity of the PMMA produced at −30 °C from 92% rr to 78% rr and 73% rr, respectively.105 The analogous monomeric magnesium alkyl, aryl, or amide complexes are also highly active catalysts for MMA polymerization, producing st-PMMA with ∼90% rr at −30 °C. Owing to the ligand-assisted, chain-end control nature of the polymerization by these discrete magnesium complexes, the syndiotacticity erodes drastically to only 75% rr for the polymerization carried out at ambient temperature (23 °C). Allen and co-workers reported in 1974 that organocalcium compounds, including (Flu)CaCl, (Ind)2Ca, Cp2Ca, and Ph2Ca, in polar DME at low temperatures converted MMA to st-PMMA; in the case of Cp2Ca, the syndiotacticity was reported to be 94% rr at Tp = 0 °C, but achieving only 8% conversion.106 This claim was later disputed by a more recent report revealing that the PMMA formed had only a modest syndiotacticity of rr = 80% when the purified Cp2Ca was used for the polymerization.107
3.3 Coordination polymerization
Similarly to the stereospecific polymerization of α-olefins, heterogeneous Ziegler–Natta type catalysts were the first transition-metal catalysts to conquer the field of stereospecific coordination polymerization of MMA. In 1965, Abe and co-workers reported the use of TiCl4/AlEt3 mixtures as effective polymerization catalysts of MMA at temperatures below 0 °C in toluene.108 It was found that Al/Ti ratios between 3–5 were optimal, and most successfully, with Al/Ti = 5 and MMA/Ti = 26 at −78 °C, the monomer conversion was 88.5% after 18 h (TOF = 1.3 h−1). The resulting PMMA had a high syndiotacticity of rr = 92% and a high MW (Table 4), giving a minimum initiator efficiency of <1%. The same authors later investigated this polymerization system in more detail and observed that the polymerization can also be mediated in a similar fashion by other trialkyl aluminum species such as triisobutylaluminum and trihexylaluminum, although there was a slight decrease in syndiotacticity (rr = 87–90).109 With AlEt3, the syndiotacticity of the resulting PMMA reported in this full paper was somewhat higher (94% rr) at Tp of −78 °C.| Catalyst | [M]/[I] | T p (°C) | TOF (h−1) | M n (kg mol−1) | PDI (Mw/Mn) | I* (%) | rr (%) | ref. |
|---|---|---|---|---|---|---|---|---|
| TiCl4/5AlEt3 | 26 | –78 | 1.3 | 255 (Mv) | n.r. | <1 | 92 | 108 |
| 28 | 500 | –78 | 26.9 | 82.0 | 1.04 | 59 | 93 | 112 |
| 28 | 1000 | –95 | 13.7 | 187 | 1.05 | 44 | 95 | 112 |
| 36 | n.r. | –78 | n.r. | 2,270 | n.r. | n.r. | 94 | 115 |
| Cp*2YbAlH3·NEt3 | 1000 | –40 | n.r. | 118 | 1.74 | 84 | 93 | 117 |
| 37 | 100 | –78 | 3 | 157 | 1.11 | 4.2 | 91 | 118 |
| 38 | 200 | –78 | 200 | 43.0 | 1.08 | 93 | 90 | 119 |
| 39/B(C6F5)3 (41) | 100 | 25 | 814 | 16.1 | 1.23 | 60 | 94 | 133 |
| 39/B(C6F5)3 (41) | 400 | 25 | 800 | 41.5 | 1.48 | 49 | 94 | 134 |
| 40/Ph3CB(C6F5)4 (42) | 100 | 25 | 394 | 20.9 | 1.37 | 45 | 94 | 133 |
| 40/Ph3CB(C6F5)4 (42) | 400 | 25 | 717 | 44.3 | 1.32 | 41 | 94 | 133 |
| 43/Ph3CB(C6F5)4 | 400 | 25 | 1,554 | 40.0 | 1.14 | 98 | 94 | 134 |
| 43/Ph3CB(C6F5)4 | 400 | 50 | 735 | n.r. | n.r. | n.r. | 93 | 134 |
| 44/Ph3CB(C6F5)4 | 400 | 25 | 1,557 | n.r. | n.r. | n.r. | 94 | 134 |
| 43/B(C6F5)3 (45) | 400 | 25 | 260 | 40.1 | 1.39 | 98 | 91 | 135 |
Yasuda's seminal work developed the highly stereospecific polymerization of MMA by organolanthanide catalysts, leading to polymers with unprecedentedly high MW's and narrow MWD's.110,111 In 1992, Yasuda and co-workers reported the use of neutral, single-component, trivalent lanthanocenes, such as dimeric samarocene hydride [Cp*2SmH]2 (28, Chart 9), as catalysts for living polymerization of MMA in toluene.112 In particular, 28 at 0 °C was shown to rapidly polymerize MMA (0.033 mol % catalyst, 3 h, 98% conversion, TOF = 980 h−1) and produce high MW PMMA (Mn = 563 kDa) with a narrow MWD (PDI = 1.04) and a moderate syndiotacticity (rr = 82.3%). Lowering Tp to −78 °C and −95 °C resulted in highly syndiotactic PMMA, with rr = 93.1% and 95.3%, respectively. Both the reactivity and catalyst efficiency are much higher than the classic heterogeneous catalyst (Table 4). It was also noted that several other analogous lanthanocenes, such as [Cp*2YMe]2 (29), Cp*2SmMe2AlMe2 (30), and Cp*2LnMe(THF) (31, Ln = Sm, Yb, Lu; Chart 10), can equally afford syndiotactic PMMA (rr > 90%) with narrow MWD's (PDI = 1.03–1.05) at low temperatures (Tp < −78 °C). Overall, these lanthanocenes function as both initiator (to effect chain initiation and growthvia conjugate Michael addition) and catalyst (to activate monomer via monomer coordination to the highly Lewis acidic Sm center) in the polymerization of MMA. Chain initiation is proposed to involve nucleophilic attack of the Sm hydride to the coordinated (activated) MMA, followed by conjugate addition of the resulting ester enolate to a second MMA coordinated to Sm, giving rise to the eight-membered-chelate propagating species (Chart 9). Propagation proceeds via repeated intramolecular conjugate Michael additions through the Sm enolate–monomer complex (active species) to the eight-membered-ring intermediate (i.e., catalyst resting state) cycle.
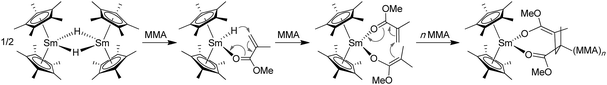 | ||
| Chart 9 Proposed chain initiation and propagation steps in the MMA polymerization by samarocene 28. | ||
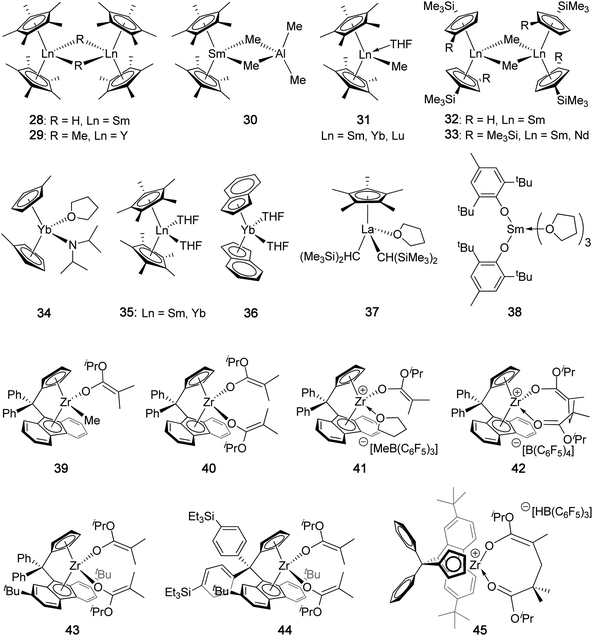 | ||
| Chart 10 Coordination catalysts that produce highly syndiotactic PMMA. | ||
Trimethylsilyl-substituted lanthanocene methyl complexes, [(Me3SiC5H4)2SmMe]2 (32) and {[(Me3Si)2C5H3]2LnMe}2 (33, Ln = Sm, Nd; Chart 10) also initiate living polymerization of MMA at −78 °C with even higher TOFs than [Cp*2SmH]2, but the PMMA syndiotacticity is somewhat lower (86–90% rr), as compared to 93% rr of the PMMA produced by [Cp*2SmH]2 at −78 °C.113 Shen et al. investigated the polymerization of (dimethylamino)ethyl methacrylate by lanthanocene amide complexes.114 In particular, this methacrylate monomer was polymerized in a syndiospecific fashion by (MeC5H4)2YbN(iPr)2(THF) (34) at −78 °C in toluene, affording a high MW (Mn = 260 kDa), syndiotactic polymer (rr = 92.8%). The catalyst had high activity with a TOF ∼ 245 h−1, but the efficiency was low with I* = 28%.
Yasuda et al. also investigated the polymerization of MMA in toluene by divalent lanthanocenes, Cp*2Ln(THF)2 (35, Ln = Sm, Yb) and (Ind)2Yb(THF)2 (36).115 All three catalysts produced polymers with narrow MWD's (PDI = 1.09–1.11) at 0 °C. At −78 °C, 36 afforded ultra high MW PMMA (Mn = 2270 kDa) with rr = 93.8%; even at −40 °C, 35-Yb afforded PMMA with rr = 91.5%. The authors noted that, in contrast to the trivalent lanthanides, all the divalent lanthanides examined afforded lower initiator efficiencies (I* < 40%). Later work by Boffa and Novak revealed that the propagating active species involved in the polymerization of MMA by divalent 35-Sm is a bimetallic samarium(III) species.116 This species was derived from a one-electron transfer from the samarium(II) complex to the monomer affording a samarium(III) cation and a MMA radical anion, which combine to form a samarium enolate radical. Two radicals then combine in a head-to-tail fashion, resulting in the bimetallic propagating species. Since two metal centers now form one polymer chain, the MW of the polymer will be at least twice as great as expected, and the observed initiator efficiency can never exceed 50% based on the unimetallic model. Knjazhanski and co-workers prepared a series of alane AlH3 complexes of divalent lanthanocenes, which produced polymers with slightly different properties than their parent, non-alane complexes.117 Specifically, Cp*2YbAlH3·NEt3 afforded PMAA with rr = 93% in toluene at −40 °C. In comparison, under otherwise identical conditions, Cp*2Yb(THF) produces PMMA with a slightly lower syndiotacticity of rr = 89%.
Half-sandwich lanthanocene complex Cp*La[CH(SiMe3)2]2(THF) (37) polymerized MMA to highly syndiotactic (91% rr), high MW (Mn = 1.57 × 105), and narrow MWD (1.11) PMMA at −78 °C, but with low activity (3 h−1TOF) and I* (∼4%).118 The polymerization at 25 °C was much more active (TOF = 200 h−1), but the syndiotacticity dropped sharply to 74% rr. A divalent non-metallocene samarium complex supported by the bulky phenoxy ligand, (BHT)2Sm(THF)3 (38), promotes living polymerization of MMA in toluene at −78 °C, producing st-PMMA (Mn = 43 KDa, PDI = 1.08, rr = 90%) with good activity (TOF = 200 h−1).119 The effect on the polymerization by adding an increasing amount of MeAl(BHT)2 was performed; increasing the [Al]/[Sm] ratio from 0 to 5 led to a decrease in rr from 90 to 20%, accompanied by an increase in mm from 1 to 68%, and a nearly constant mr.
Group IV chiral metallocenes, especially in their cationic forms, have achieved phenomenal levels of success performing as homogeneous, single-site, stereospecific catalysts for polymerization of nonpolar α-olefins to stereoregular polymers with tailored stereomicrostructures.120 Such successes have allowed for the establishment of a strong correlation between the symmetry of such chiral metallocenium catalysts and the stereomicrostructure of the resulting polymers. Thus, C2-ligated catalysts readily produce it-PP, Cs-ligated catalysts produce st-PP, C1-ligated catalysts afford either hemiisotactic or it-PP, and achiral catalysts generally produce at-PP. It seemed that, although there is a shift in mechanism from migratory insertion of α-olefins to coordinative-addition for polar conjugate olefins (acrylic monomers), such as MMA, these general rules held true, as C2-ligated metallocenium catalysts, such as {rac-[C2H4(η5-Ind)2]Zr(THF)[OC(OiPr)CMe2]}+[MeB(C6F5)3]−,121,122 at ambient temperature efficiently produce highly isotactic poly(methacrylate)s (>95% mm)121,122 and poly[(meth)acrylamide]s (>99% mm).123–127 However, when the Cs-ligated catalyst [Me2C(Cp)(Flu)ZrMe]+, which is successful in synthesizing highly syndiotactic PP,33,35,120 was used for the polymerization of MMA, no activity was observed. Although the inactivity issue was solved by using an ester enolate zirconocenium complex, {Me2C(Cp)(Flu)Zr(THF)[OC(OiPr)CMe2]}+[MeB(C6F5)3]−, the resulting PMMA was still only syndio-biased (rr = 64%).128 A different type of Cs-ligated complex incorporating a linked dianionic Cp-amido ligand, Me2Si(η5-(Me4C5)(tBuN), affords PMMA with tacticity depending on metal. Thus, the zirconium enolate complex, [Me2Si(η5-(Me4C5)(tBuN)]Zr(L)[OC(OtBu)CMe2]}+[B(ArF)4]− (ArF = 3,5-(CF3)2C6H3, L = neutral donor ligand such as THF or isobutyrate), reported by Collins et al.,129 exhibited low activity (TOF = 9.4 h−1) and afforded, unexpectedly (on the basis of its Cs ligation), highly isotactic PMMA (95.5% mm) via a site-control mechanism at low Tp (–60 °C and −40 °C) in a solvent mixture of toluene and CH2Cl2. On the other hand, Chen and co-workers found that the titanium complex bearing the same ligand set, [Me2Si(η5-(Me4C5)(tBuN)]TiMe+MeB(C6F5)3−, effected living and syndioselective polymerization at ambient temperature, producing PMMA with syndiotacticity ∼80% rr and controlled MW and narrow MWD (PDI = 1.09).130 The corresponding cationic titanium ester enolate complex, which simulates the structure of the active propagating species, behaves similarly to that of the alkyl complex, producing syndiotactic PMMA (80% rr, 18% mr, 2.0% mm) at ambient temperature with predominately isolated m meso dyad stereoerrors (…rrrrmrrrr…), again pointing to the apparent chain-end control nature of the titanium catalyst. Further studies by Chen, Cavallo and co-workers showed that catalyst site-epimerization corrects a stereomistake made in a previous enantiofacial misaddition, accounting for the formation of the predominately isolated m stereoerrors.131 Höcker and co-workers utilized the C2v-ligated complex [Me2CCp2ZrMe(THF)]+[BPh4]− to produce PMMA with rr = 89.0% in CH2Cl2 at −45 °C, via chain-end control at low temperatures.132 The catalyst activity and efficiency were low (TOF ∼ 7 h−1, I* ∼16%).
The first catalyst-site-controlled syndiospecific polymerization of MMA was reported in 2008 by Ning and Chen, enabling the synthesis of highly syndiotactic PMMA at ambient or higher temperatures.133 Neutral zirconocene mono(ester enolate) 39 and bis(ester enolate) 40 (Chart 10), supported by the rigid Ph2C< bridged Cs-symmetric ligand, Ph2C(Cp)(Flu), can be readily converted to the cationic catalystsviamethide and hydride abstraction, respectively. Specifically, activation of 39 is achieved by reaction with B(C6F5)3·THF in CH2Cl2 or toluene to cleanly generate the cationic species 41, while the activation of 40 proceeds via H− abstraction from the methyl group within the enolate moiety by Ph3C+ forming Ph3CH and isopropyl methacrylate coordinated to Zr; subsequent nucleophilic addition of another enolate ligand to this activated methacrylate monomer gives the cationic eight-membered-ring chelate 42, the active catalyst resting intermediate. Hence, the MMA polymerization at 25 °C in CH2Cl2 by bis(ester enolate) 40, upon in situactivation with Ph3CB(C6F5)4, produced highly syndiotactic PMMA (94% rr) with a high Tg of 139 °C via a predominately site-controlled mechanism. Likewise, catalyst 41, generated by in situactivation of precatalyst 39 with B(C6F5)3·THF, rapidly polymerized MMA at 25 °C in CH2Cl2 with TOF reaching 814 h−1 (95% conversion in 7 min in a [MMA]/[Zr] ratio of 100), yielding highly syndiotactic PMMA (94% rr). Catalyst 41 also polymerizes nBu methacrylate to the corresponding syndiotactic polymer (94% rr) with Mn = 34.7 kDa and PDI = 1.30. Impressively, the syndiospecficity of this catalyst system proved robust at elevated temperature, producing PMMA with rr = 92.8% even at 50 °C in toluene. However, at higher monomer-to-catalyst ratios such as 400, both catalysts failed to achieve high monomer conversions (≤50%) and also exhibited relatively low catalyst efficiencies (<50%).134 Subsequent catalyst structure-reactivity relationship studies by Chen and co-workers led to much more active and robust catalyst systems.134 In particular, catalyst [Ph2C(Cp)(2,7-tBu2-Flu)]Zr[OC(OiPr)CMe2]2 (43), when activated with Ph3CB(C6F5)4in situ, rapidly polymerized 400 equiv of MMA at 50 °C in toluene to achieve 97% conversion in 15 min, giving a high TOF of 1554 h−1. The PMMA produced exhibited a high syndiotacticity (rr = 94%) and a controlled MW (Mn = 40.0 kDa) with a narrow MWD (PDI = 1.14), thus giving rise to a nearly quantitative catalyst efficiency (I* = 98%). Unlike the chain-end control polymerization, the high level of syndiospecificity of this catalyst-site-control polymerization is not markedly altered by changing the monomer-to-catalyst ratio (up to 2000 examined), solvent and temperature (93% rr at Tp of 50 °C). Introducing substituents to the Cp ring and the Flu rings at different patterns, or substituting the Ph2C< bridge with other bridging moieties such as Me2C<, Me2Si<, or Ph2Si<, decreases the catalyst syndiospecificity, with changing the bridging exerting the most pronounced effect.134 On the other hand, placing a silyl group at the para-position of the bridging phenyl (i.e., 44) has virtually no impact on catalyst activity and syndiospecificity (44vs.43, Table 4).
Kinetic and mechanistic studies revealed that this polymerization is catalyst-site-controlled and proceeds with a monometallic, coordinative-addition mechanism, consisting of a fast intramolecular conjugate addition to form the eight-membered resting state intermediate, followed by the rate determining step (r.d.s) of opening the chelate by incoming monomer (Chart 11). Computational studies rationalized why the Ph2C< bridged catalyst exhibits higher stereoselectivity than other catalysts with the Me2C< or Me2Si< bridge. Specifically, the Ph2C< bridge rigidity pushes the η3-bound Flu ligand closer to the growing chain and the monomer, thereby increasing ΔEStereo between the competing transition states for the addition of a monomer molecule to the opposite (correct and wrong) enantiofaces of the enolate growing chain.134
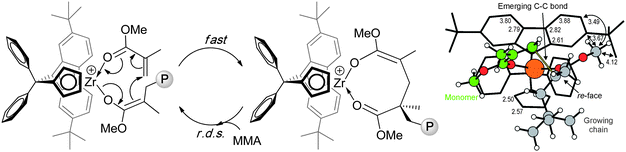 | ||
| Chart 11 Propagation “catalysis” cycle (left) and transition state generating a stereomistake in the MMA polymerization by the cationic catalyst derived from 43.134 | ||
Most recently, Chen and co-workers found that activation of Cs-ligated bis(ester enolate) metallocene precatalysts with the strong Lewis acid, B(C6F5)3, affords quantitatively the corresponding isolable cationic eight-membered ester enolate metallacycles such as 45 (Chart 10).135 This rapid, two-step reaction consists of vinylogous hydride abstraction to form the anion [HB(C6F5)3]− and nucleophilic addition of the second enolate ligand to the methacrylate resulted from loss of a hydride in the first enolate ligand to form the chelating cation. In contrast to ansa-Flu-Cp ligated, eight-membered chelating cations paired with more commonly used weakly coordinating anions, such as [MeB(C6F5)3]− or [B(C6F5)4]−, the same cations paired with the anion [HB(C6F5)3]− show drastic activity differences in different solvents, with the activity in toluene being ∼16-fold lower than that in CH2Cl2. In comparison, the [HB(C6F5)3]− based catalysts also exhibit significantly lower activity (by 6–40 fold) and produce PMMA with noticeably lower syndiotacticity. Most uniquely, [HB(C6F5)3]− based catalysts effect substantial internal chain-transfer reactions, especially for polymerizations carried out in toluene and in the presence of excess B(C6F5)3, thus releasing polymer chains with a terminal double bond and achieving a catalytic polymerization. The picture emerging from the combined experimental and theoretical study has led to a new hydride-shuttling chain transfer mechanism promoted by the hydridoborate anion, involving a hydride addition and abstraction sequence through the borane center.135
4. Poly(cyclic ester)s
4.1 Polylactide
Polylactide (PLA) is one of the most commercially important polymers that exhibits both biodegradable and biocompatible properties, enabling a wide array of applications in packaging, microelectronics, and biomedical fields.136,137PLA is conveniently synthesized by the ring opening polymerization (ROP) of the bioderived monomer, lactide.138,139 Possessing two stereocenters, lactide exists as three different diastereomers: LL- (L-LA), DD- (D-LA), and DL- (meso-LA). It has been well documented that the ROP of the enantiomeric monomers, in absence of any site-epimerization, leads to polymers with quantitative isotacticity. To synthesize syndiotactic PLA, it is required that the stereoselective ROP of meso-LA be effectively carried out, namely exhibiting a large kinetic preference for attacking one of the diastereotopic sites in meso-LA. In 1999, Ovitt and Coates utilized enantiopure chiral aluminum salen isopropoxide complex 46 for the stereoselective ROP of meso-LA, affording highly syndiotactic PLA (r % = 96%), with the annealed polymer exhibiting a Tg of 34.1 °C and Tm of 152 °C (Chart 12).140 Both Tg and Tm of st-PLA are considerably lower than those of typical it-PLA derived from the ROP of L-LA (Tg ∼ 50 °C and Tm up to 180 °C). Although the activity was low (1 mol % catalyst, 40 h, 94% conversion, TOF < 3 h−1), the polymerization was controlled, producing st-PLA with a narrow MWD of 1.05 and Mn of 12.5 kDa, giving a high initiator efficiency of I* = 88.8%. The polymerization was proposed to proceed through a catalyst site-control mechanism.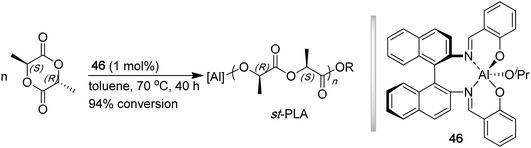 | ||
| Chart 12 Stereospecific ROP of meso-LA to synthesize st-PLA by a chiral (salen)Al catalyst. | ||
An analogous group 3 yttrium salen complex was found to be more active toward the polymerization of meso-LA, but producing only atactic PLA.140 On the other hand, by recognizing that the catalysts that polymerize rac-LA to ht-PLA should also polymerize meso-LA to st-PLA, Okuda and co-workers employed heteroselective scandium catalysts (47), containing a 1,ω-dithia-alkanediyl-bridged bis(phenolato) ligand with bulky ortho-substituents for the stereoselectiv ROP of meso-LA to highly syndiotactic PLA (Chart 13).141 The polymerizations by such catalysts supported by the tetradentate [OSSO] ligand were carried out in toluene at 25 °C, affording polymers with the highest Pr (the probability of forming a new syndiotactic, racemic dyad) approaching 0.93. These catalysts exhibit much higher activity than the above salen Al catalyst, approaching quantitative monomer conversion (1 mol % catalyst) in 30 min, giving a TOF ∼ 200 h−1. There was a great range in MWD of the polymers produced by the varying catalysts, ranging from broad (PDI = 2.15) to relatively narrow (PDI = 1.29), indicative of a varied degree of transesterification or chain transfer. The two most reactive and syndioselective catalysts with two cumyl substituents on the aromatic rings, depicted in Chart 13, also gave PLA with the measured Mn being much higher than the theoretical Mn, therefore yielding low initiator efficiencies of I* = 25–32%. The st-PLA produced has a relative low Tm of 119 °C. A structurally related indium complex supported by the tetradentate [OSSO] ligand142 also produces st-PLA (Pr = 0.93%) at room temperature, but the MWD is much narrower (PDI = 1.05).141
![Stereospecific ROP of meso-LA to st-PLA by tetradentate [OSSO]Sc catalysts.](/image/article/2011/PY/c1py00245g/c1py00245g-c13.gif) | ||
| Chart 13 Stereospecific ROP of meso-LA to st-PLA by tetradentate [OSSO]Sc catalysts. | ||
4.2 Poly(β-hydroxyalkanoate)s
Poly(3-hydroxybutyrate) (PHB), a key member of naturally occurring biodegradable and biocompatible microbial aliphatic polyesters, poly(β-hydroxyalkanoate)s (PHAs), is an isotactic polymer (Tm = 180 °C) exhibiting properties similar to crystalline thermoplastic it-PP and produced by various bacteria and algae.143,144 ROP of β-butyrolactone (βBL) provides a convenient synthetic route to high molecular weight PHB.145 While the polymerization of the enantiopure βBL by achiral or chiral initiators leads to the corresponding isotactic polymer, the polymerization of racemic βBL typically leads to an atactic polymer or a polymer with low stereoregularity. Carpentier and co-workers have developed discrete yttrium amide or alkoxide initiators (48) supported by tetradentate, dianionic amino-alkoxy-bis(phenolate) [O−,N,O,O−] ligands that promote syndiospecific polymerization of racemic βBL to syndiotactic PHB (Chart 14) by a chain-end control mechanism.146–148 The polymerization at 20 °C in toluene had living characteristics and was rapid, with TOF up to 24,000 h−1; the stereoselectivity and activity of the yttrium catalyst system can be fine-tuned by the R substituents on the phenolate ring. The resulting polymers had narrow MWD's (PDI = 1.18–1.03) and exhibited Pr as high as 0.94 (89% rr) and Tm as high 183 °C.147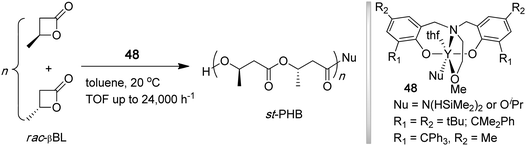 | ||
| Chart 14 Synthesis of syndiotactic PHB by living, syndiospecific ROP of rac-βBL. | ||
Thomas, Coates, and co-workers recently reported a new strategy for the synthesis of new sequentially controlled PHAs, which utilized a monomeric yttrium silylamide and a dimeric yttrium alkoxide (49, Chart 15) supported by a tetradentate phenoxyamine (salan) ligand to polymerize a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio of two different enantiomeric β-lactones with stereocenters of opposite configuration and bearing different substituents.149 Both complexes enabled full conversion of 30–500 equiv of 50:50 mixtures of β-lactones in 5–120 min, with the highest TOF reaching 4700 h−1. This polymerization system not only produced syndiotactic PHAs with narrow MWDs (PDI = 1.10–1.23 by 49), but additionally, the polymers were composed of an alternating monomer sequence (90–94% alternation). The Tm of the alternating copolymers with different β-side chains (R1 and R2) could be altered over a wide range of temperatures (47 °C to 210 °C), depending on the choice of enantiopure monomer building blocks.
:50 ratio of two different enantiomeric β-lactones with stereocenters of opposite configuration and bearing different substituents.149 Both complexes enabled full conversion of 30–500 equiv of 50:50 mixtures of β-lactones in 5–120 min, with the highest TOF reaching 4700 h−1. This polymerization system not only produced syndiotactic PHAs with narrow MWDs (PDI = 1.10–1.23 by 49), but additionally, the polymers were composed of an alternating monomer sequence (90–94% alternation). The Tm of the alternating copolymers with different β-side chains (R1 and R2) could be altered over a wide range of temperatures (47 °C to 210 °C), depending on the choice of enantiopure monomer building blocks.
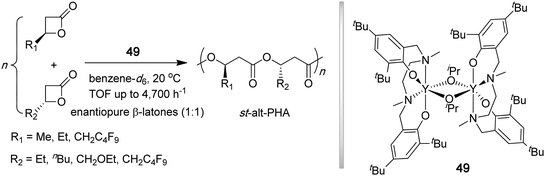 | ||
| Chart 15 Synthesis of sequence-controlled syndiotactic PHAs by alternating ROP of enantiopure β-lactones. | ||
5. Miscellaneous polymers
5.1 Higher α-olefin polymers
C s-symmetric, syndiospecific-propylene-polymerization catalysts such as 1 and its expanded derivatives have also been utilized to produce highly syndiotactic polymers derived from higher α-olefins.150 For instance, Shiomura and co-workers synthesized a series of syndiotactic poly(1-olefin)s through the syndiospecific polymerization of 1-butene, 1-pentene, 1-hexene, 4-methyl-1-pentene, and 1-octene using catalyst 1 activated with MAO at 30 °C.151 In a kinetic study Deffieux et al. utilized the 1/1000MAO system to synthesize syndiotactic (89% rr) poly(1-hexene) in toluene at 20 °C and found that the polymer syndiotacticity is substantially lowered when carrying out the polymerization in polar chlorinated solvents such as o-dichlorobenzene and methylene chloride.152 Highly syndiotactic poly(1-pentene) (rrrr > 99%, Mn = 68.4 kDa, PDI = 1.84) was synthesized by the same catalyst system (1/700MAO) in toluene at 0 °C.153 Hoff and Kaminsky synthesized syndiotactic poly(1-pentene), poly(1-hexene) and poly(1-octene) with syndiotacticity ranging from 88% to 91% rrrr using [Ph2C(Cp)(2,7-tBu2-Flu)]ZrCl2/7800MAO at Tp = 0 °C.154 Highly syndiospecific propylene polymerization catalyst 6 is also effective in the syndiospecific polymerization of 4-methyl-1-pentene; at 0 °C the neat polymerization had TOF = 4.52 × 103 h−1 and produced a syndiotactic polymer (rrrr = 97%) with a high Tm of 215 °C.39 Under similar conditions, catalyst 6 outperformed 1 in terms of syndioselectivity (TOF = 5.95 × 103 h−1, rrrr = 94%, Tm = 210 °C), while catalyst 4 was only active at 25 °C, producing an amorphous material. Zambelli et al. reported syndiospecific polymerization of β-branched 1-olefins such as (S)- and rac-4-methyl-1-hexene by 1/MAO, leading to the corresponding highly syndiotactic polymers.155,1565.2 Conjugated diolefin polymers
The stereoselective polymerization of linear conjugated dienes leads to a variety of possible stereoregular, tactic polymer structures as a result of chemo-, regio- and stereoselectivities.157 For instance, polymerization of 1,3-pentadiene (PD) can lead to one or more of the following 10 possible stereoregular polymers: 1,2-cis (iso- and syndiotactic), 1,2-trans (iso- and syndiotactic), 3,4-iso- and syndiotactic, 1,4-cis (iso- and syndiotactic), and 1,4-trans (iso- and syndiotactic) stereomicrostructures. Natta and co-workers reported in 1964 that homogeneous titanium alkoxide/aluminum alkyl catalyst systems converted 1,4-butadiene (BD) into syndiotactic 1,2-PBD (Chart 16), with concomitant formation of an amorphous polymer.158 Many other catalyst systems, predominantly cobalt-based, have subsequently been developed for the synthesis of st-1,2-PBD, with a ternary system consisting of Co(acac)3/organoaluminum/CS2 being most effective, leading to st-1,2-PBD with high 1,2-selectivity (>99%), syndiotacticity (>99%), and crystallinity (>79%, Tm up to 216 °C).159–161 Following the discovery of the syndiospecific polymerization of styrene by TiBz4 and CpTiCl3 (10) activated with MAO, the same catalyst systems were employed for the stereoselective polymerization of conjugated dienes. Zambelli,162 Oliva163 and co-workers reported the synthesis of a syndiotactic 1,2-addition polymer from the polymerization of 4-methyl-1,3-pentadiene (4MPD) by TiBz4/MAO and CpTiCl3/MAO, respectively. The syndiotactic polymer produced by TiBz4/100MAO at 20–30 °C contained ≥88% 1,2-units and is an amorphous materials, but catalytic hydrogenation afforded a fully saturated crystalline polymer with Tm = 186 °C.162 In comparison, the polymerization by CpTiCl3/1000MAO at 20 °C or −18 °C led to ≥98% 1,2-syndiotactic P4MPD (Chart 16), and the syndiotactic polymer produced at −18 °C had a Tm of 95 °C.164 Interestingly, the activity and stereoselectivity of the polymerization of 1,3-pentadiene by CpTiCl3/500MAO are highly sensitive to Tp.165 Thus, on going from Tp = 20 °C to 0 °C and −28 °C, the polymerization activity increased drastically from TOF = 15 h−1 to 73 h−1 and 154 h−1, while the resulting PPD went from being ≥99% cis-1,4-isotactic to 72% cis-1,2-syndiotactic and ≥99% cis-1,2-syndiotactic (Tm ∼ 100 °C), respectively (Chart 16). Performing the polymerization of 1,3-pentadiene by CpTiCl3/500MAO at −78 °C led to a polymer consisting exclusively of cis-1,2 units and having a syndiotactic structure, although the MW was still relatively low (Mn = 9.24 kDa and PDI = 1.7).166 The crystalline trans-1,2-st-PPD (Mn = 211 kDa, PDI = 2.4, 63% rrrr, Tm = 164 °C) was synthesized with the CoCl2(PiPrPh2)2–MAO system at −30 °C.167 Natta et al. reported in 1962 the first synthesis of crystalline cis-1,4-st-PPD (Tm = 52–53 °C) using a cobalt catalyst system consisting of Co(acac)3/AlEt2Cl/H2O–MAO.168 The water in the system is expected to react with AlEt2Cl to form an aluminoxane, the activator.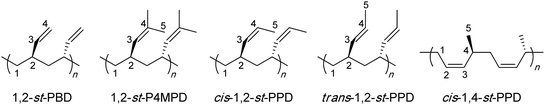 | ||
| Chart 16 Structures of syndiotactic 1,2- and 1,4-conjugated diene polymers. | ||
5.3 Bicyclic olefin polymers
Tactic polymers derived from the stereoselective ring-opening metathesis polymerization (ROMP) of bicyclic and polycyclic olefins have been recently reviewed,169 and only several more recent examples are highlighted herein. Schrock and co-workers developed syndiospecific ROMP of dicarbomethoxy norbornadiene (DCMNBD) by a Z-selective, monoaryloxide-monopyrrolide (MAP) molybdenum imidoalkylidene catalyst (2 mol% 50a), affording a highly cis (>99%), syndiotactic (>99%) ROMP polymer (Chart 17).170 Generation of such a stereomicrostructure for the ROMP polymer requires inversion at the Mo center with each forward metathesis step, namely through stereogenic catalyst-site control. Such MAP catalysts have been further utilized for the syndiospecific ROMP of 2,3-bis(trifluoromethyl)-bicyclo[2.2.1]hepta-2,5-diene and 3-methyl-3-phenylcyclopropene.171 Most recently, Schrock et al. synthesized cis,syndiotactic (>95%) ROMP polymers that incorporates alternating enantiomers through syndiospecific ROMP of racemic monomers, rac-endo-exo-5,6-dicarbomethoxynorbornene and rac-1-methyl-5,6-dicarbomethoxy-7-oxanorbornadiene, using 1 mol% MAP Mo catalyst 50b (Chart 17).172 | ||
| Chart 17 Synthesis of cis, syndiotacticROMP polymers containing alternating enantiomers. | ||
5.4 (Meth)acrylamide polymers
Despite the structural similarities between (meth)acrylates and (meth)acrylamides and the successes in synthesizing highly isotactic polyacrylamides,173,174 there have been very few reports on the syndiospecific polymerization of (meth)acrylamides. Okamoto and co-workers reported the syndiospecific radical polymerization of N-methyl methacrylamide.175 Through screening a wide range of radical initiators and polymerization conditions such as solvent, temperature, and concentration, the authors found optimal conditions for high syndiospecificity. Thus, employing the nBu3B-air initiating system and performing the polymerization in (CF3)2CHOH at −78 °C with a [M]/[I] ratio of 50 resulted in only 7% polymer yield after 24 h, but the resulting polymer was highly syndiotactic (rr = 95%) and had Mn = 8 kDa and PDI = 2.4. Okamoto et al. carried out a similar study for the radical polymerization of N,N-dimethylacrylamide (DMAA) and N,N-diphenylacrylamide (DPAA) in fluoroalchols.176 Interestingly, the radical polymerization of DMAA resulted in isotactic-biased polymers, whereas under similar polymerization conditions, polymerization of DPAA resulted in syndiotactic-biased polymers. On the other hand, the radical polymerization initiated by nBu3B-air, at −78 °C or −98 °C in THF, afforded highly syndiotactic PDPAA, with rr = 91% and 93%, respectively. The synthesis of highly syndiotactic PDMAA at much higher temperature (room temperature) has been recently achieved by catalyst-site-controlled coordination polymerization with Cs-ligated metallocenium catalysts.1346. Summary and outlook
As can be readily seen from the above survey, there has been an intense pursuit in the synthesis of highly syndiotactic polymers from a variety of monomer substrates. Among several mechanisms by which polymerization proceeds to yield highly syndiotactic polymers, coordination polymerization catalyzed by discrete metal complexes offers the capability for such synthesis to be carried out under economically viable conditions (e.g., industrial scale production of st-PP and st-PS). This trait is due to the ability of coordination polymerization to control the polymerization characteristics, especially its stereochemistry, at ambient and above temperatures, often in a catalytic fashion, by either site-control with chiral catalysts or chain-end control with steric interplay between the catalyst ligand, the coordinated monomer and the growing-chain end. It is also readily seen that the typical activity achieved for the coordination-insertion polymerization of nonpolar α-olefins is higher than that observed for the coordination-addition polymerization of polar conjugated olefins (acrylic monomers) by several orders of magnitude. Furthermore, the latter polymerization is typically carried out at low temperatures to achieve high syndiospecificity, until very recently, and is rarely catalytic. It is also important to recognize the fact that renewable monomer substrates derived directly from biomass are oxygenated, polar feedstocks. Hence, future research in this field will be largely directed at addressing these currently unmet challenges in the stereoselective polymerization of polar monomers, by developing stereoselective catalysts that can render high-speed coordination-insertion or -addition polymerization of polar feedstocks in a catalytic fashion, and exploring the synthesis of environmentally sustainable polymers, especially stereoregular ones, from renewable resources.Acknowledgements
The authors are grateful for the financial support provided by the U. S. National Science Foundation (NSF-1012326).References
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- D. Baskaran and A. H. E. Müller, Prog. Polym. Sci., 2007, 32, 173–219 CrossRef CAS.
- D. Baskaran, Prog. Polym. Sci., 2003, 28, 521–581 CrossRef CAS.
- K. Hatada, T. Kitayama and K. Ute, Prog. Polym. Sci., 1988, 13, 189–276 CrossRef CAS.
- W. A. Braunecker and Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- S. Yamago, Chem. Rev., 2009, 109, 5051–5068 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS.
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- H. Yasuda, Prog. Polym. Sci., 2000, 25, 573–626 CrossRef CAS.
- L. S. Baugh and J. A. M. Canich, ed., Stereoselective Polymerization with Single-Site Catalysts, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008 Search PubMed.
- L. Del Villano, M. A. Kelland, G. M. Miyake and E. Y.-X. Chen, Energy Fuels, 2010, 24, 2554–2562 CrossRef CAS.
- K. Ute, N. Miyatake and K. Hatada, Polymer, 1995, 36, 1415–1419 CrossRef CAS.
- A. D. Bolig and E. Y.-X. Chen, J. Am. Chem. Soc., 2002, 124, 5612–5613 CrossRef CAS.
- A. Rodriguez-Delgado, W. R. Mariott and E. Y.-X. Chen, Macromolecules, 2004, 37, 3092–3100 CrossRef CAS.
- A. D. Bolig and E. Y.-X. Chen, J. Am. Chem. Soc., 2001, 123, 7943–7944 CrossRef CAS.
- P. Galli and G. Vecellio, J. Polym. Sci., Part A: Polym. Chem., 2003, 42, 396–415.
- J. Severn and R. L. Jones Jr., in Handbook of Transition Metal Polymerization Catalysts, ed. R. Hoff and R. T. Mathers, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, pp. 157–230 Search PubMed.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1345 CrossRef CAS.
- A. Razavi, V. Bellia, Y. De Brauwer, K. Hortmann, L. Peters, S. Sirole, S. Van Belle and U. Thewalt, Macromol. Chem. Phys., 2004, 205, 347–356 CrossRef CAS.
- J. Schellenberg, Syndiotactic Polystyrene: Synthesis, Characterization, Processing, and Application, John Wiley and Sons, Inc., Hoboken, New Jersey, 2010 Search PubMed.
- J. Schellenberg, Prog. Polym. Sci., 2009, 34, 688–718 CrossRef CAS.
- A. Rodrigues, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2008, 252, 2115–2136 CrossRef CAS.
- N. Tomotsu, N. Ishihara, T. H. Newman and M. T. Malanga, J. Mol. Catal. A: Chem., 1998, 128, 167–190 CrossRef CAS.
- G. Natta, J. Polym. Sci., 1955, 16, 143–154 CrossRef CAS.
- N. Ishihara, M. Kuramoto and M. Uoi, JP 62187708, 1985.
- N. Ishihara, T. Seimiya, M. Kuramoto and M. Uoi, Macromolecules, 1986, 19, 2464–2465 CrossRef.
- N. Ishihara, M. Kuramoto and M. Uoi, Macromolecules, 1988, 21, 3356–3360 CrossRef CAS.
- C. De Rosa and F. Auriemma, Prog. Polym. Sci., 2006, 31, 145–237 CrossRef CAS.
- J. A. Ewen, R. L. Jones, A. Razavi and J. D. Ferrara, J. Am. Chem. Soc., 1988, 110, 6255–6256 CrossRef CAS.
- A. Razavi and J. D. Ferrara, J. Organomet. Chem., 1992, 435, 299–310 CrossRef CAS.
- A. Razavi and J. L. Atwood, J. Organomet. Chem., 1993, 459, 117–123 CrossRef CAS.
- D. Veghini, L. M. Henling, T. J. Burkhards and J. E. Bercaw, J. Am. Chem. Soc., 1999, 121, 564–573 CrossRef CAS.
- A. Razavi, V. Bellia, Y. De Brauwer, K. Hortmann, L. Peters, S. Sirole, S. Van Belle and U. Thewalt, Macromol. Chem. Phys., 2004, 205, 347–356 CrossRef CAS.
- S. A. Miller and J. E. Bercaw, Organometallics, 2004, 23, 1777–1789 CrossRef CAS.
- L. J. Irwin and S. A. Miller, J. Am. Chem. Soc., 2005, 127, 9972–9973 CrossRef CAS.
- Z. Cai, T. Ikeda, M. Akita and T. Shiono, Macromolecules, 2005, 38, 8135–8139 CrossRef CAS.
- T. A. Herzog, D. L. Zubris and J. E. Bercaw, J. Am. Chem. Soc., 1996, 118, 11988–11989 CrossRef CAS.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS.
- M. Mitani, R. Furuyama, J.-I. Mohri, J. Saito, S. Ishii, H. Terao, T. Nakano, H. Tanaka and T. Fujita, J. Am. Chem. Soc., 2003, 125, 4293–4305 CrossRef CAS.
- J. Tian and G. W. Coates, Angew. Chem., Int. Ed., 2000, 39, 3626–3629 CrossRef CAS.
- J. Tian, P. D. Hustad and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 5134–5135 CrossRef CAS.
- M.-C. Chen, J. A. S. Roberts and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 4605–4625 CrossRef.
- H. Ma and J. Huang, in Stereoselective Polymerization with Single-Site Catalysts, ed. L. S. Baugh and J. A. M. Canich, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008, ch. 14, pp. 363–397 Search PubMed.
- J. E. Mark, Polymer Data Handbook, Oxford University Press, New York, 1999 Search PubMed.
- J. Schellenberg and H.-J. Leder, Adv. Polym. Technol., 2006, 25, 141–151 CrossRef CAS.
- M. Malanga, Adv. Mater., 2000, 12, 1869–1872 CrossRef CAS.
- W. Kaminsky, S. Lenk, V. Scholz, H. W. Roesky and A. Herzog, Macromolecules, 1997, 30, 7647–7650 CrossRef CAS.
- C. Pellecchia, P. Longo, A. Proto and A. Zambelli, Makromol. Chem. Rapid Commun., 1992, 13, 265–268 CrossRef CAS.
- A. Grassi, C. Lamberti, A. Zambelli and I. Mingozzi, Macromolecules, 1997, 30, 1884–1889 CrossRef CAS.
- C. Pellecchia, D. Pappalardo, L. Oliva and A. Zambelli, J. Am. Chem. Soc., 1995, 117, 6593–6594 CrossRef CAS.
- H. Kucht, A. Kucht, J. C. W. Chien and M. D. Rausch, Appl. Organomet. Chem., 1994, 8, 393–394 CrossRef CAS.
- T. E. Ready, R. O. Day, J. C. W. Chien and M. D. Rausch, Macromolecules, 1993, 26, 5822–5823 CrossRef CAS.
- T. E. Ready, J. C. W. Chien and M. D. Rausch, J. Organomet. Chem., 1996, 519, 21–28 CrossRef CAS.
- P. Poster, J. C. W. Chien and M. D. Rausch, Organometallics, 1996, 15, 2404–2409 CrossRef CAS.
- N. Schneider, M.-H. Prosence and H.-H. Brintzinger, J. Organomet. Chem., 1997, 545–546, 291–295 CrossRef CAS.
- G. Xu and E. Ruckenstein, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2481–2488 CrossRef CAS.
- S. Y. Knjazhanski, G. Moreno, G. Cadenas, V. K. Belsky and B. M. Bulychev, Tetrahedron, 1999, 55, 1639–1646 CrossRef CAS.
- S. Y. Knazhanski, G. Cadenas, M. García and C. M. Pérez, Organometallics, 2002, 21, 3094–3099 CrossRef CAS.
- E. Kirillov, C. W. Lehmann, A. Razavi and J.-F. Carpentier, J. Am. Chem. Soc., 2004, 126, 12240–12241 CrossRef CAS.
- Y. Sarazin, P. de Frémont, L. Annunziata, M. Duc and J.-F. Carpentier, Adv. Synth. Catal., 2011, 353, 1367–1374 CrossRef CAS.
- A.-S. Rodrigues, E. Kirillov, C. W. Lehmann, T. Roisnel, B. Vuillemin, A. Razavi and J.-F. Carpentier, Chem.–Eur. J., 2007, 13, 5548–5565 CrossRef.
- Y. Luo, J. Baldmus and Z. Hou, J. Am. Chem. Soc., 2004, 126, 13910–13911 CrossRef CAS.
- F. Bonnet, C. D. C. Violante, P. Roussel, A. Mortreux and M. Visseaux, Chem. Commun., 2009, 3380–3382 RSC.
- Y. Luo, X. Feng, Y. Wang, S. Fan, J. Chen, Y. Lei and H. Liang, Organometallics, 2011, 30, 3270–3274 CrossRef CAS.
- T. Miyatake, K. Mizunuma and M. Kakugo, Makromol. Chem., Macromol. Symp., 1993, 66, 203–214 Search PubMed.
- J. Okuda and E. Masound, Macromol. Chem. Phys., 1998, 199, 543–545 CAS.
- J. C. W. Chien, Z. Salajka and S. Dong, Macromolecules, 1992, 25, 3199–3203 CrossRef CAS.
- A. Grassi, A. Zambelli and F. Laschi, Organometallics, 1996, 15, 480–482 CrossRef CAS.
- A. Grassi, S. Saccheo and A. Zambelli, Macromolecules, 1998, 31, 5588–5591 CrossRef CAS.
- A. Grassi, C. Pellecchia, L. Oliva and F. Laschi, Macromol. Chem. Phys., 1995, 196, 1093–1100 CrossRef CAS.
- M. K. Mahanthappa and R. M. Waymouth, J. Am. Chem. Soc., 2001, 123, 12093–12094 CrossRef CAS.
- P. Longo, A. Proto and A. Zambelli, Macromol. Chem. Phys., 1995, 196, 3015–3029 CrossRef CAS.
- A. Zambelli, C. Pellecchia and A. Proto, Macromol. Symp., 1995, 89, 373–382 CrossRef CAS.
- L. Perrin, E. Kirillov, J.-F. Carpentier and L. Maron, Macromolecules, 2010, 43, 6330–6336 CrossRef CAS.
- J. B. Lando, J. Semen and B. Farmer, Macromolecules, 1970, 3, 524–527 CrossRef CAS.
- C. Chovino and P. Gramain, Macromol. Chem. Phys., 1996, 197, 1411–1418 CrossRef CAS.
- Y. Isobe, K. Yamada, T. Nakano and Y. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4693–4703 CrossRef CAS.
- Y. Miura, T. Satoh, A. Narumi, O. Nishizawa, Y. Okamoto and T. Kakuchi, Macromolecules, 2005, 38, 1041–1043 CrossRef CAS.
- T. Serizawa, K. Hamada and M. Akashi, Nature, 2004, 429, 52–55 CrossRef CAS.
- H. Ishizawa, T. Nakano, T. Yade, M. Tsuji, O. Nakagawa and T. Yamaguchi, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4656–4665 CrossRef CAS.
- D. L. Glusker, R. A. Galluccio and R. A. Evans, J. Am. Chem. Soc., 1964, 86, 187–196 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2004, pp 640–643, 699–701 Search PubMed.
- K. Hatada and T. Kitayama, Polym. Int., 2000, 49, 11–47 CrossRef CAS.
- P. Vlček and L. Lochmann, Prog. Polym. Sci., 1999, 24, 793–973 CrossRef CAS.
- C. Zune and R. Jérôme, Prog. Polym. Sci., 1999, 24, 631–664 CrossRef CAS.
- T. Kitayama, T. Shinozaki, E. Masuda, M. Yamamoto and K. Hatada, Polym. Bull., 1988, 20, 505–510 CAS.
- T. Kitayama, T. Shinozaki, T. Sakamoto, M. Yamamoto and K. Hatada, Makromol. Chem., 1989, 15, 167–185 CrossRef CAS.
- T. Kitayama, T. Hirano, Y. Zhang and K. Hatada, Macromol. Symp., 1996, 107, 297–306 CrossRef CAS.
- T. Kitayama, T. Hirano and K. Hatada, Tetrahedron, 1997, 53, 15263–15279 CrossRef CAS.
- T. Kitayama, S. He, Y. Hironaka, T. Iijima and K. Hatada, Polym. J., 1995, 27, 314–318 CrossRef CAS.
- D. Kunkel, A. H. E. Müller, M. Janata and L. Lochmann, Makromol. Chem., Macromol. Symp., 1992, 60, 315–326 Search PubMed.
- D. Seebach, Angew. Chem., Int. Ed. Engl., 1988, 27, 1624–1654 CrossRef.
- A. Rodriguez-Delgado and E. Y.-X. Chen, J. Am. Chem. Soc., 2005, 127, 961–974 CrossRef CAS.
- E. Y.-X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3395–3403 CrossRef CAS.
- R. Kakuchi, K. Chiba, K. Fuchise, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 8747–8750 CrossRef CAS.
- T. Kitayama, E. Masuda, M. Yamaguchi, T. Nishiura and K. Hatada, Polym. J., 1992, 24, 817–827 CrossRef CAS.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS.
- Y. Joh and Y. Kotake, Macromolecules, 1970, 3, 337–345 CrossRef CAS.
- K. Hatada, H. Nakanishi, K. Ute and T. Kitayama, Polym. J., 1986, 18, 581–591 CrossRef CAS.
- A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Williams, Chem. Commun., 2002, 1208–1209 RSC.
- E. L. Marshall and V. C. Gibson, in Stereoselective Polymerization with Single-Site Catalysts, ed. L. S. Baugh and J. A. M. Canich, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008, ch. 23, pp. 593–644 Search PubMed.
- K. A. Allen, B. G. Gowenlock and W. E. Lindsell, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 1131–1133 CrossRef CAS.
- Y. Li, H. Deng, W. Brittain and M. S. Chisholm, Polym. Bull., 1999, 42, 635–639 CrossRef CAS.
- H. Abe, K. Imai and M. Matsumoto, J. Polym. Sci., Part B: Polym. Lett., 1965, 3, 1053–1058 CrossRef CAS.
- H. Abe, K. Imai and M. Matsumoto, J. Polym. Sci., Part C, 1968, 23, 469–485 Search PubMed.
- H. Yasuda, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1955–1959 CrossRef CAS.
- H. Yasuda and E. Ihara, Adv. Polym. Sci., 1997, 133, 53–101 CrossRef CAS.
- H. Yasuda, H. Yamamoto, K. Yokota, S. Miyake and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 4908–4910 CrossRef CAS.
- Y. Satoh, N. Ikitake, Y. Nakayama, S. Okuno and H. Yasuda, J. Organomet. Chem., 2003, 667, 42–52 CrossRef CAS.
- Q. Shen, Y. Wang, K. Zhang and Y. Yao, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 612–616 CrossRef CAS.
- H. Yasuda, H. Yamamoto, M. Yamashita, K. Yokota, A. Nakamura, S. Miyake, Y. Kai and N. Kanehisa, Macromolecules, 1993, 26, 7134–7143 CrossRef CAS.
- L. S. Boffa and B. M. Novak, Macromolecules, 1994, 27, 6993–6995 CrossRef CAS.
- S. Y. Knjazhanski, L. Elizalde, G. Cadenas and B. M. Bulychev, J. Organomet. Chem., 1998, 568, 33–40 CrossRef CAS.
- K. Tanaka, M. Furo, E. Ihara and H. Yasuda, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1382–1390 CrossRef CAS.
- M. Nodono, T. Tokimitsu and T. Makino, Macromol. Chem. Phys., 2003, 204, 877–884 CrossRef CAS.
- L. Resconi, J. C. Chadwick and L. Cavallo, in Comprehensive Organomettalic Chemistry III, ed. M. Bochmann, chief. ed. M. P. Mingos and R. H. Crabtree, Elseier, Oxford, 2007, vol. 4, pp. 1005–1166 Search PubMed.
- A. Rodriguez-Delgado and E. Y.-X. Chen, Macromolecules, 2005, 38, 2587–2594 CrossRef CAS.
- A. D. Bolig and E. Y.-X. Chen, J. Am. Chem. Soc., 2004, 126, 4897–4906 CrossRef CAS.
- G. M. Miyake, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Macromolecules, 2009, 42, 1462–1471 CrossRef CAS.
- G. M. Miyake and E. Y.-X. Chen, Macromolecules, 2008, 41, 3405–3416 CrossRef CAS.
- G. M. Miyake, W. R. Mariott and E. Y.-X. Chen, J. Am. Chem. Soc., 2007, 129, 6724–6725 CrossRef CAS.
- W. R. Mariott and E. Y.-X. Chen, Macromolecules, 2005, 38, 6822–6832 CrossRef CAS.
- W. R. Mariott and E. Y.-X. Chen, Macromolecules, 2004, 37, 4741–4743 CrossRef CAS.
- A. Rodriguez-Delgado and E. Y.-X. Chen, J. Organomet. Chem., 2006, 691, 3490–3497 CrossRef CAS.
- H. Nguyen, A. P. Jarvis, M. J. G. Lesley, W. M. Kelly, S. S. Reddy, N. J. Taylor and S. Collins, Macromolecules, 2000, 33, 1508–1510 CrossRef CAS.
- A. Rodriguez-Delgado, W. R. Mariott and E. Y.-X. Chen, Macromolecules, 2004, 37, 3092–3100 CrossRef CAS.
- Y. Ning, L. Caporaso, A. Correa, L. O. Gustafson, L. Cavallo and E. Y.-X. Chen, Macromolecules, 2008, 41, 6910–6919 CrossRef CAS.
- H. Frauenrath, H. Keul and H. Höcker, Macromolecules, 2001, 34, 14–19 CrossRef CAS.
- Y. Ning and E. Y.-X. Chen, J. Am. Chem. Soc., 2008, 130, 2463–2465 CrossRef CAS.
- Y. Zhang, Y. Ning, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2010, 132, 2695–2709 CrossRef CAS.
- Y. Zhang, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2011, 133, 1572–1588 CrossRef CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hederick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- J.-C. Buffet, A. Kapelski and J. Okuda, Macromolecules, 2010, 43, 10201–10203 CrossRef CAS.
- I. Peckermann, A. Kapelski, T. P. Spaniol and J. Okuda, Inorg. Chem., 2009, 48, 5526–5534 CrossRef CAS.
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1–8 CrossRef CAS.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem.–Eur. J., 2011, 17, 1872–1883 CrossRef CAS.
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS.
- M. Kol, S. Segal and S. Groysman, in Stereoselective Polymerization with Single-Site Catalysts, ed. L. S. Baugh and J. A. M. Canich, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008, ch. 13, pp. 345–361 Search PubMed.
- T. Asanuma, Y. Nishimori, M. Ito, N. Uchikawa and T. Shiomura, Polym. Bull., 1991, 25, 567–570 CrossRef CAS.
- D. Coevoet, H. Cramail and A. Deffieux, Macromol. Chem. Phys., 1999, 200, 1208–1214 CrossRef CAS.
- V. Grumel, R. Brüll, H. Pasch, H. G. Raubenheimer, R. Sanderson and U. M. Wahner, Macromol. Mater. Eng., 2002, 287, 559–564 CrossRef CAS.
- M. Hoff and W. Kaminsky, Macromol. Chem. Phys., 2004, 205, 1167–1173 CrossRef CAS.
- A. Zambelli, A. Grassi, M. Galimberti and G. Perego, Makromol. Chem. Rapid Commun., 1992, 13, 269–275 CrossRef CAS.
- A. Zambelli, A. Grassi, M. Galimberti and G. Perego, Makromol. Chem. Rapid Commun., 1992, 13, 467–469 CrossRef CAS.
- A. Proto and C. Capacchione, in Stereoselective Polymerization with Single-Site Catalysts, ed. L. S. Baugh and J. A. M. Canich, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008, ch. 17, pp. 447–487 Search PubMed.
- G. Natta, L. Porri and A. Carbonaro, Makromol. Chem., 1964, 77, 126–138 CrossRef CAS.
- H. Ashitaka, H. Ishikawa, H. Ueno and A. Nagasaka, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1853–1860 CrossRef CAS.
- H. Ashitaka, K. Jinda and H. Ueno, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1951–1972 CrossRef CAS.
- H. Ashitaka, K. Inaishi and H. Ueno, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1973–1988 CrossRef CAS.
- A. Zambelli, P. Ammendola and A. Proto, Macromolecules, 1989, 22, 2126–2128 CrossRef CAS.
- L. Oliva, P. Longo, A. Grassi, P. Ammendola and C. Pellecchia, Makromol. Chem. Rapid Commun., 1990, 11, 519–524 CrossRef CAS.
- G. Ricci, S. Italia, A. Giarrusso and L. Porri, J. Organomet. Chem., 1993, 451, 67–72 CrossRef CAS.
- G. Ricci, S. Italia and L. Porri, Macromolecules, 1994, 27, 868–869 CrossRef CAS.
- G. Ricci, E. Alberti, L. Zetta, T. Motta, F. Bertini, A. Mendichi, P. Arosio, A. Famulari and S. V. Meille, Macromolecules, 2005, 38, 8353–8361 CrossRef CAS.
- G. Ricci, A. Forni, A. Boglia, T. Motta, G. Zannoni, M. Canetti and F. Bertini, Macromolecules, 2005, 38, 1064–1070 CrossRef CAS.
- G. Natta, L. Porri, A. Carbonaro, F. Ciampelli and G. Allegra, Makromol. Chem., 1962, 51, 229–232 CrossRef CAS.
- D. Breen, K. Curran and W. Risse, in Stereoselective Polymerization with Single-Site Catalysts, ed. L. S. Baugh and J. A. M. Canich, CRC Press/Taylor & Francis Group, Boca Raton, Florida, 2008, ch. 20, pp. 509–551 Search PubMed.
- M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS.
- M. M. Flook, L. C. H. Gerber, G. T. Debelouchina and R. R. Schrock, Macromolecules, 2010, 43, 7515–7522 CrossRef CAS.
- M. M. Flook, V. W. L. Ng and R. R. Schrock, J. Am. Chem. Soc., 2011, 133, 1784–1786 CrossRef CAS.
- Y. Okamoto, K. Suzuki, K. Ohta, K. Hatada and H. Yuki, J. Am. Chem. Soc., 1979, 101, 4763–4765 CrossRef CAS.
- T. Nakano, Y. Okamoto and K. Hatada, J. Am. Chem. Soc., 1992, 114, 1318–1329 CrossRef CAS.
- J. Zhang, W. liu, T. Nakano and Y. Okamoto, Polym. J., 2000, 32, 694–699 CrossRef CAS.
- W. Liu, T. Nakano and Y. Okamoto, Polym. J., 2000, 32, 771–777 CrossRef CAS.
Footnote |
| † Current address: Division of Chemistry and Chemical Engineering, Caltech. |
| This journal is © The Royal Society of Chemistry 2011 |