Applying “click” chemistry to polyurethanes: a straightforward approach for glycopolymer synthesis†
Christina
Ott
*a,
Christopher D.
Easton
a,
Thomas R.
Gengenbach
a,
Sally L.
McArthur
b and
Pathiraja A.
Gunatillake
*a
aCSIRO Materials Science and Engineering, Bag 10, Clayton South MDC, Victoria 3169, Australia. E-mail: Christina.Ott@csiro.au; Thilak.Gunatillake@csiro.au; Fax: +61 3 9545 2446; Tel: +61 3 9545 2369
bBiointerface Engineering Group, IRIS, Swinburne University of Technology, PO Box 218, Hawthorn, Victoria 3122, Australia
First published on 21st October 2011
Abstract
We report a facile synthetic route to prepare polyurethanes with pendant sugar-moieties in the side-chain of the polymer through incorporation of diverse chain extenders capable of undergoing either copper catalyzed Huisgen 1,3-dipolar cycloaddition or thiol-ene click reactions.
Since the click chemistry concept was introduced by Sharpless and co-workers1 in 2001, many academic and industrial scientists have widely explored this innovative approach for various research areas2 as a result of its versatility, efficiency and inertness towards functional groups. The click reaction is commonly performed in the presence of a copper catalyst which may represent a limitation with respect to applications in biological3 systems due to associated toxicity factors. Therefore, researchers are constantly seeking to develop highly efficient and orthogonal reactions which do not require any metal catalyst for in vivo biomedical applications. Recently, Schubert et al. published an excellent overview of the latest achievements in metal-free click reactions.4 A variety of alternative synthetic approaches are now considered as “click” reactions, particularly when certain requirements are fulfilled such as: modularised methodology that is wide in scope, readily available starting materials, simple reaction conditions, regioselectivity, quantitative yields and facile purification procedures. Many of the above mentioned criteria apply to the radical addition reaction between thiols and C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.5 One key advantage of this reaction is its broad applicability in material synthesis. The thiol-ene reaction has been proven to be a powerful synthetic tool for the design of complex macromolecular architectures. For example, Lowe and co-workers have described a facile convergent approach to synthesize 3-arm star polymers and telechelic polymers by using a RAFT6homopolymer capable of undergoing thiol-ene as well as thiol-yne click reactions.7 Moreover, the synthetic potential has also been exploited for the construction of well-defined dendrimers (Hawker et al.).8
C double bonds.5 One key advantage of this reaction is its broad applicability in material synthesis. The thiol-ene reaction has been proven to be a powerful synthetic tool for the design of complex macromolecular architectures. For example, Lowe and co-workers have described a facile convergent approach to synthesize 3-arm star polymers and telechelic polymers by using a RAFT6homopolymer capable of undergoing thiol-ene as well as thiol-yne click reactions.7 Moreover, the synthetic potential has also been exploited for the construction of well-defined dendrimers (Hawker et al.).8
Polyurethanes (PUs) remain an essential class of synthetic polymers. These materials are in high demand due to their versatility and ability to tune the material properties by the choice and composition of the starting materials. Areas of application include the automotive, electrical/electronics and construction industries. In addition, tremendous progress has been made in the field of biomaterials where PUs play a decisive role in the development of medical devices and implants due to their favorable bio- and blood-compatibility properties. Recent research has focused on synthetic strategies to attach biorecognizable groupse.g.carbohydrates capable of specifically interacting with cell surface receptor proteins.9 Davis et al. described the functionalization of RAFT6-derived copolymers with glucose using a thio-halogen click reaction as well as a nucleophilic substitution based on a para-toluenesulfonyl precursor.10 Another post-modification reaction with glucose was performed on pentafluorostyrene copolymers which readily react with thiol-functionalized molecules by selectively replacing the para-fluorine groups.11 Du Prez et al.12 and Cho et al.13 have recently demonstrated the feasibility of performing copper-catalyzed Huisgen 1,3-dipolar cycloaddition reactions in the side-chain of PUs. Surprisingly, this approach has not yet been applied for the modification of PUs with carbohydrates. Herein, we report an efficient synthetic route for preparing novel glycopolymers by combining polyurethane chemistry with a subsequent functionalization of the synthesized materials via “click” procedures. For this purpose, carefully chosen chain extenders possessing alkene and alkyne functionalities, respectively, were applied for step-growth polymerization processes. Depending on the utilized chain extender, modification of the polymers was achieved by performing either copper catalyzed Huisgen 1,3-dipolar cycloaddition or thiol-ene click reactions. To the best of our knowledge, this is the first metal free click modification applied to PUs.
The alkyne and alkene-functionalized polyurethanes were prepared using the bulk polymerization method, i.e. without the use of any solvent. For this purpose, 0.52 equivalents of the chain extender DPPD or AOPD (Fig. 1), 0.52 equivalents of the macrodiol poly(tetramethylene oxide) (PTMO), 1 equivalent of hexamethylene diisocyanate (HDI) and the catalyst dibutyltin dilaurate were added to a beaker and mixed thoroughly until the mixture became highly viscous. Scheme 1 outlines the general synthetic approach to functionalized polyurethanes.
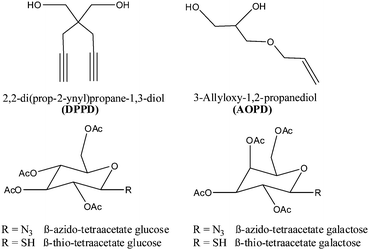 | ||
| Fig. 1 Chain extenders and β-azido/thio-tetraacetate sugars used for the polycondensation process and the subsequent modification reaction. | ||
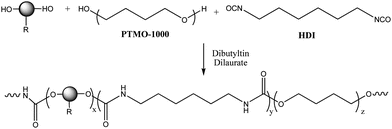 | ||
| Scheme 1 Schematic representation of the polyurethanes synthesized using differently functionalized chain extenders. | ||
After curing (5 h, 75 °C), the polymers were characterized by 1H NMR spectroscopy and GPC. The 1H NMR spectra of the corresponding PUs reveal the successful incorporation of the chain extender due to the clearly resolved alkenyl and alkynyl signals between 5–6 ppm and 2–2.5 ppm, respectively (Fig. 2).
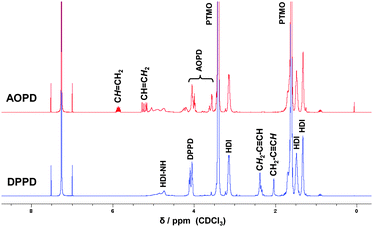 | ||
| Fig. 2 1H NMR spectra of the polyurethanes prepared with the chain extenders DPPD and AOPD. | ||
Broad molecular weight distributions (Mn ∼ 40.000 g mol−1) with polydispersity indices ranging from 1.7 to 2.7 were obtained by GPC measurements. These are expected values for polymers prepared by step-growth polymerization processes. Analysis of the DPPD-PU by IR-spectroscopy indicates the presence of alkyne function due to the peak at 3330 cm−1. In addition, three intense bands corresponding to the amide functionality were observed at 1714 cm−1 (CO stretch), 1518 cm−1 (C–N stretch), and 1235 cm−1 (C–N deformation).
The postmodification of the PU side-chains with β-azido-tetraacetate glucose (and galactose) was performed at 60 °C in DMSO using Cu(I)Br and PMDETA as catalytic system. The final synthetic step involved the cleavage of the acetate groups. For this purpose, the polymer was dissolved in chloroform and sodium methoxide in methanol was added dropwise to the mixture. After 1 h stirring at room temperature the solution was concentrated and precipitated into ice-cold diethyl ether. The disappearance of the acetyl groups (at around 2 ppm) was confirmed by 1H NMR spectroscopy as it is shown for the galactose-functionalized PU in Fig. 3.
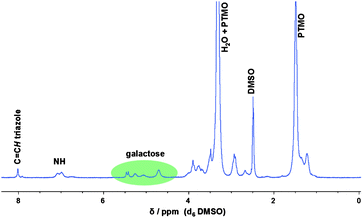 | ||
| Fig. 3 1H NMR spectra of the polyurethane modified with galactose after applying copper-catalyzed “click” chemistry. | ||
Furthermore, signals belonging to the galactose moiety between 4.5 to 5.5 ppm as well as the characteristic signal for the proton linked to the formed triazole ring were observed. Moreover, the methylene signal adjacent to the alkyne group (around 2.1 ppm) completely disappeared and shifted downfield which is evidence for qualitative conversion of the click reaction. The requirement for using the prepared glycopolymers as biomaterials is the entire removal of the copper catalyst. ICP-AES measurements revealed a copper content of 29 ppm after only two treatments with an ion exchange resin. However, as a significant amount of amide groups are present in the polymer backbone which are able to complex copper ions, a different synthetic strategy (based on hydrothiolation) was developed to avoid the usage of metal catalysts.
Initially, a model reaction was carried out to investigate the feasibility of the reaction with thio-functionalized sugars. Hence, β-thio-tetraacetate glucose was dissolved in chloroform and reacted with 4-bromo-butene in the presence of IRGACURE 651 as photoinitiator. The reaction mixture was irradiated at 365 nm for 20 min. The crude mixture was investigated by 1H NMR spectroscopy proving the success of the reaction. Afterwards, the alkene-functionalized PU was reacted with the thio-functionalized sugars using the same reaction conditions yielding PUs with glucose as well as galactose moieties in the side-chain of the polymer. Fig. 4 demonstrates the rapid and highly efficient progress of the thiol-ene reaction and consequently confirms unambiguously the structure of both polymers.
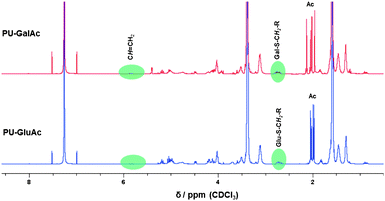 | ||
| Fig. 4 1H NMR spectra of the glucose and galactose-modified polyurethanes using thiol-ene chemistry. | ||
Both spectra indicate the disappearance of the original peaks belonging to the alkene-functionalized PU at 5.8 ppm. Furthermore, the signals between 3.7 and 5.4 ppm are attributed to the incorporated sugar moiety; their respective thiol-peaks from the starting material at 2.3 ppm are no longer present. The new signal at 2.7 ppm can be assigned to the methylene group next to the thioether functionality. The acetyl protecting groups were removed using sodium methoxide in the same manner as it was described for the two DPPD-based glycopolymers. IR spectroscopy measurements were performed for the AOPD-PU, the glucose and galactose modified PUs. However, the only observable difference was an increase in intensity of the band between 3800 and 3000 cm−1 (OH and CH stretch region) which can be attributed to the incorporated carbohydrate moieties. Due to the fact that the PUs contained only 35% hard segment, i.e. weight ratio of chain extender and diisocyanate to macrodiol, the characteristic bands for the alkenyl C–H and CC stretch could not be observed in the spectrum of AOPD-PU. Fig. 5 shows the results of X-ray photoelectron spectroscopy (XPS) investigations which were performed to follow the modification of the polymer. The polymers were dissolved in chloroform and drop-casted onto glass slides. After washing the samples with Milli-Q water, they were dried in an oven overnight. High resolution spectra of the carbon signal (C 1s) shows the functional groups within the polymer with the following assignments: 285 eV – C–C, C–H; 286.5 eV – C–O–C; 289.5 eV – NHC(O)O. Comparing both spectra, it can be noted that the peak assigned to ether groups has a greater intensity for the glycopolymer as a result of the incorporated sugar moieties; additional evidence for the effective modification via the thiol-ene reaction.
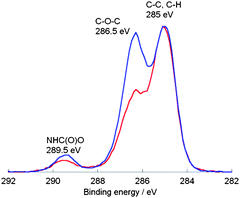 | ||
| Fig. 5 C 1s high resolution XPS spectra for the AOPD-PU (red) and the glucose-modified PU (blue). | ||
Water contact angle measurements were performed on spin-casted films to evaluate the hydrophilicity change of the polymer before and after the modification reaction. The contact angle decreased from 89° (AOPD-PU) to 72° (Glu/Gal-AOPD-PU) which can be explained by the incorporation of the hydrophilic carbohydrate groups. An example of contact angle measurement images is presented in Fig. 6.
 | ||
| Fig. 6 Contact angle measurement images of AOPD-PU (left) and glucose-modified PU (right). | ||
Conclusions
Herein is described a facile and efficient synthetic approach to synthesize novel glycopolymers by step-growth polymerization. Key to this strategy is the incorporation of pendant alkyne and alkene functional groups into PUs by using accordingly functionalized diol chain extenders. The feasibility to modify the chain extenders by copper catalyzed Huisgen 1,3-dipolar cycloaddition and thiol-ene click reactions has been verified by several analytical techniques. To the best of our knowledge, this is the first report using “click” chemistry strategies to incorporate carbohydrate moieties into PUs. In addition, it is the first time that metal free “click” reactions were employed in polyurethane chemistry. The attachment of biologically active species is of especial interest because it may procure and improve the acceptance and healing of biomedical devices or implants. Future work of these materials will include cell viability and proliferation studies.The authors are grateful to the CSIRO Office of the Chief Executive (OCE) Science Team for financial support.
Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13 CrossRef CAS; J. K. Chen, Nat. Chem. Biol., 2008, 4, 391 CrossRef; Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef; J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639 CrossRef.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS; G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959 RSC; B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef CAS; R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415 CrossRef.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS.
- I. Pashkuleva and R. L. Reis, J. Mater. Chem., 2010, 20, 8803 RSC; S. G. Spain and N. R. Cameron, Polym. Chem., 2011, 2, 60 RSC.
- C. Boyer, A. Bousquet, J. Rondolo, M. R. Whittaker, M. H. Stenzel and T. P. Davis, Macromolecules, 2010, 43, 3775 CrossRef CAS.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387 CrossRef CAS.
- D. Fournier and F. Du Prez, Macromolecules, 2008, 41, 4622 CrossRef CAS; L. Billiet, D. Fournier and F. Du Prez, Polymer, 2009, 50, 3877 CrossRef.
- S. Rana, S. Y. Lee and J. W. Cho, Polym. Bull., 2010, 64, 401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details. See DOI: 10.1039/c1py00412c |
| This journal is © The Royal Society of Chemistry 2011 |