DOI:
10.1039/C1RA00007A
(Paper)
RSC Adv., 2011,
1, 135-141
Oligostilbenes from Vatica mangachapoi with xanthine oxidase and acetylcholinesterase inhibitory activities†
Received
31st March 2011
, Accepted 7th April 2011
First published on 20th July 2011
Abstract
Phytochemical examination of the branches and twigs of the tropical plant Vatica mangachapoi resulted in the isolation of three novel oligostilbenes named vaticahainols A–C (1–3) along with seventeen known oligostilbenes (4–20). The structures of the new compounds were elucidated on the basis of extensive spectroscopic data analysis, modified Mosher's method, and quantum chemical computation. All of the isolated phytochemicals were screened for their xanthine oxidase (XO) and acetylcholinesterase (AChE) inhibitory activities.
Introduction
Dipterocarpaceous plants are well known as rich sources of various resveratrol (3,5,4′-trihydroxystilbene) oligomers,1 many of which exhibit remarkable bioactivities such as antitumor,2,3antioxidant,4 antibacterial,5,6 immunosuppressive7 and AChE inhibitory8,9 effects. Vatica, a genus comprising 65 species, belongs to the largest subfamily Dipterocarpoideae in the Dipterocarpaceae, most of which are tall, hardy and evergreen trees primarily distributed in Southeast Asia.
In the course of our ongoing search for new and/or bioactive agents of tropical plant origins,2,8 an ethyl acetate extract of the branches and twigs of V. mangachapoi, collected in Hainan island, China, was considered to be a potent inhibitor of XO and AChE. V. mangachapoi, a rather rare species only occur in Hainan island in China, has not been investigated previously and therefore was subjected to detailed laboratory investigation. We report herein the detailed isolation, structural elucidation and bioassay of these polyphenols.
Results and discussion
Bioassay-guided fractionation of the EtOAc-soluble extract of the branches and twigs of V. mangachapoi led to the isolation of three novel stilbenoid dimers (1–3) and seventeen known oligostilbenes (4–20) (Fig. 1). The structures of the new compounds were established by spectroscopic data interpretation, modified Mosher's method and quantum chemical computation. Moreover, other known compounds were identified as vaticanol A (4),10 vaticanol E (5),11 pauciflorol B (6),12 vatdiospyroidol (7),13 diptoindonesin D (8),14(+)-parviflorol (9),15(+)-ε-viniferin (10),16(+)-ampelopsin F (11),17 isoampelopsin F (12),18 hopeafuran (13),19(+)-ampelopsin A (14),20 hopeahainol D (15),4 pauciflorol C (16),12 hemsleyanol D (17),21 hopeaphenol A (18),22 vaticaffinol (19),23 and davidol A (20)24 by appropriate literature consultations.
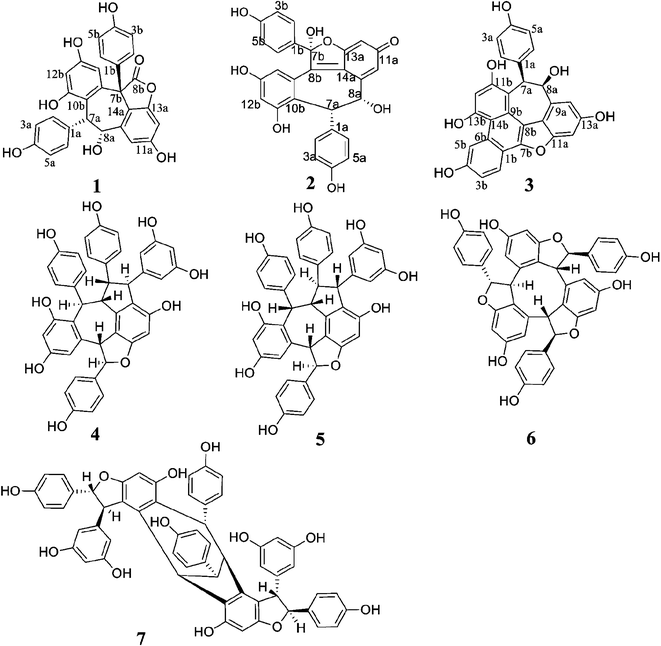 |
| | Fig. 1 Structures of oligostilbenes isolated from Vatica mangachapoi. | |
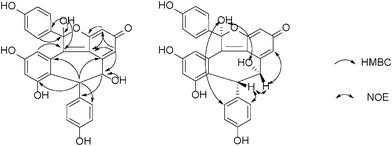
Vaticahainol A (1) was obtained as a white amorphous powder. Its molecular formula was established as C28H20O8 on the basis of a quasimolecular ion peak at m/z 485.1230 [M+H]+ ([C28H21O8]+, calc. 485.1231) in its HRESI mass spectrum. The IR spectrum of 1 displayed absorption bands at 3271–3394 cm−1 indicative of OH groups, as well as those at 1775, 1594 and 1513 cm−1 characteristic of phenyl and ester ketone moieties. The 1H NMR, HMQC and 1H–1H COSY spectra showed the presence of two 4-hydroxyphenyl groups [δ 6.52 (2H, d, J = 8.2 Hz, H-2a/H-6a), 6.05 (2H, d, J = 8.2 Hz, H-3a/H-5a); and 6.15 (4H, brs, H-2b/H-6b, H-3b/H-5b)], two 1,2,3,5-tetrasubstituted benzene rings [δ 7.08 (1H, J = 1.5 Hz, H-10a), 6.56 (1H, J = 1.5 Hz, H-12a); 6.44 (1H, d, J = 2.0 Hz, H-12b), and 7.25 (1H, d, J = 2.0 Hz, H-14b)], and a set of mutually coupled aliphatic protons [δ 5.22 (1H, d, J = 4.0 Hz) and 4.67 (1H, t, J = 4.0 Hz)]. The spectra further showed signals due to an aliphatic hydroxyl group [δ 5.55 (1H, d, J = 4.0 Hz)] coupled with H-8a and five phenolic hydroxyl groups (δ 8.60–10.05). Their positions on four aromatic rings were established by analysis of correlations in the HMBC spectrum. In the HMBC spectrum, the correlations between H-7a/C-2a(6a), H-7a/C-9b and C-11b, H-8a/C-10a and C-14a, H-14b/C-7b, and H-2b(6b)/C-7b revealed the linkages of C-1a/C-7a, C-7a/C-10b, C-8a/C-9a, C-7b/C-9b and C-1b/C-7b, respectively. Considering the molecular formula, the remaining two oxygen, one carbon atoms and two unsaturated degrees were attributed to a lactone (δC-8b = 175.7) ring, anchored at C-7b (δ 57.9) and C-13a (δ 152.7) to form a γ-butalactone. The planar structure of vaticahainol A, a rearranged stilbene dimer, was thus determined.
The absolute configuration of 1 was determined by a combination of NOESY spectrum, modified Mosher's method, and quantum chemical computation (Fig. 2). The NOE correlation of H-7a/H-8a together with the lack of any correlations between the proton pairs of H-8a/H-2a(6a) and H-7a/H-10a suggested that H-7a and H-8a were cis-oriented.13 To carry out the desired Mosher reaction, compound 1 was methylated with MeI to afford a pentamethyl ether (1a) leaving a single secondary hydroxyl group at C-8a, which was further treated with R-(−)- and S-(+)-MTPA-Cl to yield S-(−)- and R-(+)-MTPA esters 1b and 1c, respectively (Scheme 1). The ΔδS-R value distribution pattern clearly indicated an 8aR configuration (Fig. 3). To assign the absolulte stereochemistry of C-7b, a comparison was made between the experimental CD and the theoretical ECD spectra for (7bS)-1 and (7bR)-1. As shown in Fig. 4, the calculated ECD curve for (7bS)-1 agreed well with the experimental ECD spectrum, which suggested that the absolute configuration of C-7b was S, the same as its biosynthetic congener hopeahainol A.8 Thus the absolute configuration of 1 was established as shown in Fig. 1.
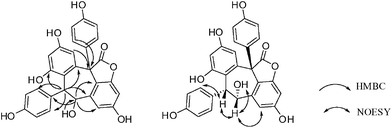 |
| | Fig. 2 Selected HMBC and NOE correlations of 1. | |
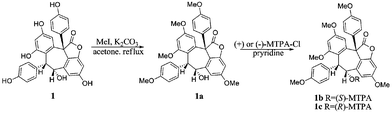 |
| | Scheme 1 | |
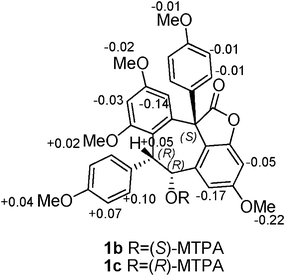 |
| | Fig. 3
ΔδS-R values (ppm) of MTPA-esters 1b and 1c. | |
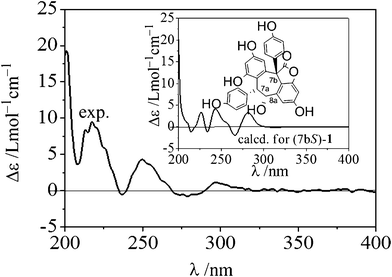 |
| | Fig. 4 The experimental and calculated ECD spectra (as shown in the inset) of (7bS)-1 after a UV correction of 11 nm. The calculation was carried out at the B3LYP/6-31G(d) level in the polarizable continuum model (CH3OH solvent: dielectric constant ε = 32.63). | |
Vaticahainol B (2), a yellow amorphous powder, gave an [M+H]+ ion peak at m/z 485.1232 in positive ion HRESIMS corresponding to the molecular formula C28H20O8, suggesting that 2 was a resveratrol dimer. The 1H NMR spectrum of 2 revealed the presence of two sets of ortho-coupled aromatic protons. They were assignable to two 4-hydroxyphenyl groups [δ 6.65 (2H, d, J = 8.5 Hz, H-2a/H-6a), 6.43 (2H, d, J = 8.5 Hz, H-3a/H-5a); 6.95 (2H, d, J = 8.5 Hz, H-2b/H-6b), 6.63 (2H, d, J = 8.5 Hz, H-3b/H-5b)]. However, only one 1,2,3,5-tetrasubstituted benzene ring [δ 6.44 (1H, d, J = 2.0 Hz, H-12b) and 7.23 (1H, d, J = 2.0 Hz, H-14b)] was revealed from its 1H NMR and 1H–1H COSY spectra. The presence of the other six-membered ring system was apparent from the signals of C-9a and C-14a in the 13C NMR spectrum (Table 1). The ring was composed of four quaternary sp2 C-atoms (δ 146.6, 188.6, 171.0, 124.1), and two protonated sp2 C-atoms (δ 99.4 and 126.1), which suggested that the ring formed a cyclohexa-2,5-dienone system. This ring system was also supported by the cross-peaks of H-10a/C-14a, H-12a/C-14a, and H-10a/C-12a in the HMBC spectrum. Further analysis of the 1H NMR, HMQC and 1H–1H COSY spectra showed a set of mutually coupled aliphatic protons at δ 5.10 (1H, brs, H-7a) and 4.81 (1H, brs, H-8a), four phenolic hydroxyl groups (δ 9.04–9.67) and one aliphatic hydroxyl, which were coupled with H-8a. The HMBC correlations of H-7a/C-2a(6a), H-7a/C-9b and C-11b, H-10a/C-8a, H-14b/C-8b, and H-2b(6b)/C-7b revealed the linkages of C-1a/C-7a, C-7a/C-10b, C-8a/C-9a, C-8b/C-9b, and C-1b/C-7b, respectively. The hydroxyl singlet at δ 8.67 showed HMBC correlations with C-1b, C-7b and C-8b revealed the aliphatic OH group located at C-7b and the linkage sequence of C-1b/C-7b/C-8b. Moreover, the remaining oxygen atom was attributable to an ether bond between C-7b and C-13a, deduced from the chemical shift values of δC-7b 116.3 and δC-13a 171.0. The planar structure of 2 was determined as a stilbene dimer with a unusual quinone ring.
Table 1
1H and 13C NMR data for compounds 1–3
| Position |
1
a
|
Position |
2
a
|
Position |
3
b
|
|
δ
C
|
δ
H (J in Hz) |
δ
C
|
δ
H (J in Hz) |
δ
C
|
δ
H (J in Hz) |
|
Measured at 500 MHz for 1H NMR and at 125 MHz for 13C NMR in DMSO-d6.
Measured at 400 MHz for 1H NMR and at 100 MHz for 13C NMR in acetone-d6.
|
| 1a |
130.3 |
|
1a |
130.5 |
|
1a |
133.0 |
|
|
2a,6a
|
129.1 |
6.52(d, 8.2) |
2a,6a
|
130.7 |
6.65(d, 8.5) |
2a,6a
|
130.5 |
6.83(d, 8.8) |
|
3a,5a
|
113.5 |
6.05(d, 8.2) |
3a,5a
|
115.8 |
6.43(d, 8.5) |
3a,5a
|
114.2 |
6.29(d, 8.8) |
| 4a |
153.9 |
|
4a |
159.4 |
|
4a |
155.4 |
|
| 7a |
44.0 |
5.22(d, 4.0) |
7a |
45.9 |
5.10(s) |
7a |
48.4 |
5.82(d, 1.8) |
| 8a |
71.1 |
4.67(t, 4.0) |
8a |
73.5 |
4.81(s) |
8a |
73.9 |
5.51(d, 1.8) |
| 9a |
143.1 |
|
9a |
146.6 |
|
9a |
139.1 |
|
| 10a |
109.7 |
7.08(d, 1.5) |
10a |
126.1 |
6.26(s) |
10a |
115.2 |
|
| 11a |
158.4 |
|
11a |
188.6 |
|
11a |
157.3 |
|
| 12a |
96.0 |
6.56(d, 1.5) |
12a |
99.4 |
5.39(d, 1.5) |
12a |
95.9 |
6.90(dd, 1.2,2.0) |
| 13a |
152.7 |
|
13a |
171.0 |
|
13a |
155.8 |
|
| 14a |
114.4 |
|
14a |
124.1 |
|
14a |
109.0 |
7.11(dd, 1.2,2.0) |
| 1b |
131.1 |
|
1b |
130.4 |
|
1b |
115.1 |
|
|
2b,6b
|
127.1 |
6.15(brs) |
2b,6b
|
129.0 |
6.95(d, 8.5) |
2b |
121.4 |
8.17(d, 8.8) |
|
3b,5b
|
114.6 |
6.15(brs) |
3b,5b
|
116.1 |
6.63(d, 8.5) |
3b |
114.9 |
7.21(dd, 8.8,2.0) |
| 4b |
155.9 |
|
4b |
157.6 |
|
4b |
156.4 |
|
| 7b |
57.9 |
|
7b |
116.3 |
|
5b |
114.1 |
9.39(d, 2.0) |
| 8b |
175.7 |
|
8b |
152.9 |
|
6b |
133.3 |
|
| 9b |
137.8 |
|
9b |
132.6 |
|
7b |
151.0 |
|
| 10b |
118.2 |
|
10b |
121.7 |
|
8b |
114.3 |
|
| 11b |
157.4 |
|
11b |
157.0 |
|
9b |
131.8 |
|
| 12b |
101.9 |
6.44(d, 2.0) |
12b |
107.6 |
6.44(d, 2.0) |
10b |
115.3 |
|
| 13b |
155.8 |
|
13b |
157.0 |
|
11b |
154.7 |
|
| 14b |
105.8 |
7.25(d, 2.0) |
14b |
111.3 |
7.23(d, 2.0) |
12b |
101.7 |
6.96(s) |
|
OH-8a
|
|
5.55(d, 4.0) |
OH-8a
|
|
6.15(d, 4.0) |
13b |
156.8 |
|
|
OH-7b
|
|
|
OH-7b
|
|
8.67(s) |
14b |
111.3 |
|
|
OH
|
|
8.60(brs) |
OH
|
|
9.04(brs) |
|
|
|
| |
|
9.11(brs) |
|
|
9.43(brs) |
|
|
|
| |
|
9.42(brs) |
|
|
9.59(brs) |
|
|
|
| |
|
9.64(brs) |
|
|
9.67(brs) |
|
|
|
| |
|
10.05(brs) |
|
|
|
|
|
|
The relative stereochemistry of the two chiral carbon atoms (C-7a/C-8a) could be determined as cis by NOE correlations between H-7a/H-8a and no correlations between H-8a/H-2a(6a) (Fig. 5). The NOE correlations of 7b-OH/H-2a(6a) and 8a-OH suggested that 7b-OH and H-7a/H-8a were trans-oriented. The absolute stereochemistry was also determined by comparing the experimental and theoretical electronic circular dichroism (ECD) spectra. The calculated ECD curve for (7aR, 8aR, 7bR)-2 agreed well with the experimentally measured spectrum (Fig. 6). Thus, the absolute configuration of 2 was 7aR, 8aR, 7bR.
 |
| | Fig. 5 Selected HMBC and NOE correlations of 2. | |
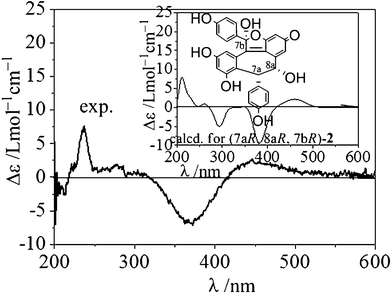 |
| | Fig. 6 The experimental and calculated ECD spectra of (7aR, 8aR, 7bR)-2. The calculation was carried out at the B3LYP/6-31G(d) level in the polarizable continuum model (CH3OH solvent: dielectric constant ε = 32.63). | |
Vaticahainol C (3), isolated as a yellow amorphous powder, had the molecular formula C28H18O7 as indicated by its HRESIMS of the [M+H]+ ion peak at m/z 467.1124 ([C28H19O7]+, calc. 467.1125). The IR spectrum showed absorption bands for hydroxyl and phenyl moieties at 3208 and 1612, 1511 cm−1, respectively. The 1D and 2D NMR spectra showed the presence of two ortho-coupled aromatic signals at δ 6.83 (2H, d, J = 8.8 Hz, H-2a/H-6a) and 6.29 (2H, d, J = 8.8 Hz, H-3a/H-5a) assignable to a para-hydroxyphenyl group; a 1,2,4-trisubstituted benzene ring protons at δ 9.39 (1H, d, J = 2.0 Hz, H-5b), 8.17 (1H, d, J = 8.8 Hz, H-2b), and 7.21 (1H, dd, J = 8.8, 2.0 Hz, H-3b); a 1,2,3,5-tetrasubstituted benzene ring proposed from a set of meta-coupled aromatic proton doublets at δ 6.90 (1H, dd, J = 1.2, 2.0 Hz, H-12a), 7.11 (1H, dd, J = 1.2, 2.0 Hz, H-14a); an aromatic proton at δ 6.96 (1H, s, H-12b); and a set of mutually coupled aliphatic protons at δ 5.82 (1H, d, J = 1.8 Hz, H-7a) and 5.51 (1H, d, J = 1.8 Hz, H-8a). In the HMBC spectrum, cross-peaks observed between H-5b/C-3b, C-14b, as well as H-2b/C-4b, C-6b, C-7b, H-12b/C-10b, C-14b and H-3b/C-1b revealed the presence of a phenanthrene moiety formed by the cyclization of the resveratrol unit, as supported by the fact that the proton signal of H-5b (δ 9.39) appeared at a relatively lower field, which was characteristic of the H-5 of a phenanthrene structure.25HMBC correlations of H-2a(6a)/C-1a, C-7a indicated that the para-hydroxyphenyl group was located at C-7a. In addition, the connections between C-7a with C-8a and C-10b were demonstrated by the correlations of H-7a with C-8a, C-9a, C-9b, C-10b and C-11b. The spectrum also displayed significant correlations between H-8a/C-9a, C-10a, C-14a, which confirmed the linkage between C-8a/C-9a. The planar structure of vaticahainol C was thus determined as a stilbene dimer with a unique phenanthrene group.
The relative stereochemistry of 3 was assigned on the basis of ROESY experiments (Fig. 7). The correlations of H-7a/H-8a, H-7a/H-2a (6a) and the lack of any correlations between the proton pairs of H-8a/H-2a(6a) suggested that H-7a and H-8a were situated in a cis-orientation to each other. A comparison was also made between the experimental CD and calculated ECD spectra for (7aR, 8aR)-3 (Fig. 8). The calculated ECD curve for (7aR, 8aR)-3 was in well agreement with the experimentally measured spectrum. Thus, the absolute configurations of C-7a and C-8a were R and R, respectively.
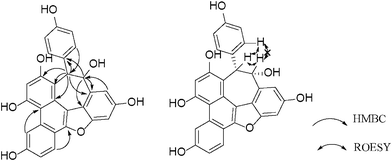 |
| | Fig. 7 Selected HMBC and ROESY correlations of 3. | |
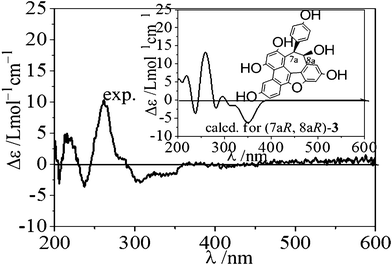 |
| | Fig. 8 The experimental and calculated ECD spectra of (7aR, 8aR)-3. The calculation was carried out at the B3LYP/6-31G(d) level in the polarizable continuum model (CH3OH solvent: dielectric constant ε = 32.63). | |
Therapeutic approaches for gout treatment include the use of XO inhibitors, which block the terminal step of uric acid synthesis. Allopurinol, the only inhibitor of the enzyme in clinical use, however, can result in a number of adverse side effects.26AChE inhibitors can be used as an effective clinical approach to treat Alzheimer's disease (AD) and the search for safer and more effective AChE inhibitors is still in great demand for the treatment of AD.27 The enzymatic inhibition rates of all the isolated oligostilbenes are shown in Table 2 and compared with those of the positive controls. Compound 4 showed inhibitory activity against XO, with an IC50 value of 23.3 ± 1.8 μM, while other compounds were almost inactive (IC50 > 50 μM). Compounds 2, 5, 6 and 7 exhibited significantly higher activities against AChE, compared to the rest of the isolates (IC50 > 40 μM), with IC50 value of 18.9 ± 1.7, 12.0 ± 1.4, 10.9 ± 1.2 and 7.3 ± 1.8 μM, respectively. The difference in inhibition rates between compounds 4 and 5 indicated that the correct stereochemistry is important for an efficient inhibition. Compound 1, although containing the 4b-OH and the lactone ring, which were supposed to explain the potent activity of hopeahainol A (IC50 = 4.33 ± 0.17 μM),8 had weak activity presumably because of the different solid geometry interfered the binding with AChE. It should be highlighted that the activity of compound 7 was close to that of the positive control (±)-huperzine A (IC50 = 1.7 ± 0.3 μM), a clinical anti-Alzheimer drug.
Table 2 Inhibition of XO and AChE by isolated oligostilbenes and positve controls
| Compound |
Enzyme (IC50/μM)a |
|
XO
|
AChE
|
|
IC50 value represents the concentration of compounds required to inhibit the 50% activity of the enzymes. “±” represents the standard deviation of the mean of these enzyme inhibition assays. Allopurinol and (±)-huperzine A were used as positive controls.
|
| 2 |
— |
18.9 ± 1.7 |
| 4 |
23.3 ± 1.8 |
— |
| 5 |
— |
12.0 ± 1.4 |
| 6 |
— |
10.9 ± 1.2 |
| 7 |
— |
7.3 ± 1.8 |
|
Allopurinol
|
5.9 ± 0.8 |
— |
| (±)-Huperzine A |
— |
1.7 ± 0.3 |
Conclusions
To the best of our knowledge there has been no prior report on the phytochemical constitution of V. mangachapoi. From this study, all isolated phytochemicals were oligostilbenes, and three of them were novel. Vaticanol A (4) showed a moderate XO inhibitory activity, while AChE inhibitory activity was obtained for vaticahainol B (2), vaticanol E (5), pauciflorol B (6) and vatdiospyroidol (7). The inhibitory potency and natural availability of these polyphenols could warrant further studies to assess their potential as effective natural remedies.
Experimental
General experimental procedures
Xanthine oxidase (XO), xanthine, electric-eel AChE (EC 3.1.1.7), acetylthiocholine iodide (ATCI), 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB) were purchased from Sigma-Aldrich Chemicals, Shanghai, P. R. China. All other chemicals used in the study were of analytical grade. Acetone-d6 and DMSO-d6 for NMR measurements were purchased from Merck, Inc. Optical rotations were taken on a Rudolph Autopol III automatic polarimeter. Fourier transform NMR spectra were recorded at 25 °C at 500/400 (for 1H) and 125/100 MHz (for 13C) on a Bruker DRX-500/400 NMR spectrometer in acetone-d6 or DMSO-d6 with tetramethylsilane (TMS) or solvent signals as internal references. UV spectra were recorded on a Hitachi U-3000 spectrophotometer in MeOH. IR spectra were measured on a Nexus 870 FT-IR spectrometer. CD spectra were acquired on a JASCO J-810 spectrometer. HRESIMS were recorded on an Agilent 6210 TOF LC/MS equipped with an electrospray ionization (ESI) probe operating in positive or negative ion mode. The semi-preparative HPLC was accomplished over an Elite ODS column (250 × 10 mm, 5 μm, Elite Scientific Co., China) on a Hitachi HPLC system consisted of an L-7110 pump (Hitachi) with an L-7110 UV-VIS Detector (Hitachi). Analytical TLC was performed on GF254 (10–20 mm) plates with 0.2 mm layer thickness. Column chromatography (CC) was fulfilled on silica gel (200–300 mesh), reverse phase (ODS) silica gel (12 nm, S-50 μm, YMC, Japan) or Sephadex LH-20 (GE, USA). Silica gel (200–300 mesh) and GF254 (10–20 mm) were produced by Qingdao Marine Chemical Factory, Qingdao, P. R. China.
Plant material
V. mangachapoi was collected in July 2003 from the Botanical Garden affiliated with the South China University of Tropical Agriculture with the specimen identified by Prof. X. Q. Zheng at Hainan University with a voucher specimen (IFB030733) deposited therein.
The air-dried twigs and branches of V. mangachapoi (7.8 kg) were powdered and soaked in EtOH (5 × 10 L) at room temperature for 7 days. In vacuoevaporation of the solvent from the extract gave a residue (188.0 g), which was suspended in H2O (500 mL) followed by sequential partitions with petroleum benzine (3 × 500 mL), EtOAc (3 × 500 mL) and n-butanol (3 × 500 mL) to afford the petroleum benzine- (5 g), EtOAc- (125.8 g) and n-butanol-soluble (11.0 g) fractions after evaporating the solvent under reduced pressure. The EtOAc extract (shown to be rich in polyphenols) was chromatographed on a silica gel column (800 g, 200–300 mesh) eluted with a gradient of CHCl3-MeOH mixtures (v/v 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 100:5, 100:10, 100:15, 100:20, 100:30, 100:50 and 0:100, each 3.5 L) to afford ten fractions A–J, following TLC monitoring. Repeated column chromatographies of fraction C over silica gel and Sephadex LH-20 followed by the semi-preparative HPLC (MeOH/H2O: 45/55, 2 ml/min) gave 1 (206.0 mg, tR = 8.3 min), 10 (304.2 mg, tR = 15.9 min) and 13 (505.5 mg, tR = 12.4 min). Fraction D was separated into ten fractions D1–D10 over a silica gel column eluting with CHCl3–MeOH mixtures of an increasing polarity (100:0–100:20). Gel filtration of D2 over a Sephadex LH-20 column with MeOH afforded 5 (1.03 g), and the work-up of D3 in the same manner supplied 8 (957.6 mg) and 9 (845.2 mg) in addition to two mixtures D3-1 and D3-2. D3-1 was further refined over a silica gel column eluting with CHCl3–MeOH (100:10) to give 12 (1.40 g), and chromatography of D3-2 over a reversed-phase silica gel column eluting with a MeOH–H2O gradient (20%→100% MeOH) afforded 2 (32.4 mg) and 15 (914.8 mg). Chromatography of D4 over silica gel column utilizing CHCl3/MeOH mixtures (100:10, 100:15, 100:20, 0:100) furnished 11 (67.4 mg) besides two sub-fractions D4-1 and D4-2. HPLC separation of D4-2 with MeOH/H2O (43/57, 2 ml/min) gave 3 (20.7 mg, tR = 10.6 min) and 14 (510.4 mg, tR = 12.5 min). Fraction E was chromatographed over a silica gel column using CHCl3–MeOH mixtures (100:0→100:30) to give three sub-fractions E1–E3. Gel filtration of E2 over Sephadex LH-20 column with MeOH afforded 4 (566.4 mg) and 6 (1.03 g). Fraction F was separated by reversed-phase silica gel column eluting with MeOH–H2O (20%→100% MeOH) to give four mixtures F1–F4. Gel filtration of F1 over Sephadex LH-20 with MeOH furnished 16 (412.5 mg) and 19 (1.52 g). Compounds 17 (525.6 mg), 18 (1.03 g) and 7 (825.6 mg) were obtained from F2 using silica gel column eluting with CHCl3/MeOH (100:15, 100:20, 100:30), followed by gel filtration over Sephadex LH-20. And F3 was chromatographed over a Sephadex LH-20 column with MeOH supplied 20 (1.29 g).
:0, 100:5, 100:10, 100:15, 100:20, 100:30, 100:50 and 0:100, each 3.5 L) to afford ten fractions A–J, following TLC monitoring. Repeated column chromatographies of fraction C over silica gel and Sephadex LH-20 followed by the semi-preparative HPLC (MeOH/H2O: 45/55, 2 ml/min) gave 1 (206.0 mg, tR = 8.3 min), 10 (304.2 mg, tR = 15.9 min) and 13 (505.5 mg, tR = 12.4 min). Fraction D was separated into ten fractions D1–D10 over a silica gel column eluting with CHCl3–MeOH mixtures of an increasing polarity (100:0–100:20). Gel filtration of D2 over a Sephadex LH-20 column with MeOH afforded 5 (1.03 g), and the work-up of D3 in the same manner supplied 8 (957.6 mg) and 9 (845.2 mg) in addition to two mixtures D3-1 and D3-2. D3-1 was further refined over a silica gel column eluting with CHCl3–MeOH (100:10) to give 12 (1.40 g), and chromatography of D3-2 over a reversed-phase silica gel column eluting with a MeOH–H2O gradient (20%→100% MeOH) afforded 2 (32.4 mg) and 15 (914.8 mg). Chromatography of D4 over silica gel column utilizing CHCl3/MeOH mixtures (100:10, 100:15, 100:20, 0:100) furnished 11 (67.4 mg) besides two sub-fractions D4-1 and D4-2. HPLC separation of D4-2 with MeOH/H2O (43/57, 2 ml/min) gave 3 (20.7 mg, tR = 10.6 min) and 14 (510.4 mg, tR = 12.5 min). Fraction E was chromatographed over a silica gel column using CHCl3–MeOH mixtures (100:0→100:30) to give three sub-fractions E1–E3. Gel filtration of E2 over Sephadex LH-20 column with MeOH afforded 4 (566.4 mg) and 6 (1.03 g). Fraction F was separated by reversed-phase silica gel column eluting with MeOH–H2O (20%→100% MeOH) to give four mixtures F1–F4. Gel filtration of F1 over Sephadex LH-20 with MeOH furnished 16 (412.5 mg) and 19 (1.52 g). Compounds 17 (525.6 mg), 18 (1.03 g) and 7 (825.6 mg) were obtained from F2 using silica gel column eluting with CHCl3/MeOH (100:15, 100:20, 100:30), followed by gel filtration over Sephadex LH-20. And F3 was chromatographed over a Sephadex LH-20 column with MeOH supplied 20 (1.29 g).
Vaticahainol A (1).
White amorphous powder; UV (MeOH): λmax (log ε): 223 (5.2), 255 (5.1), 402 (5.0) nm; IR (KBr) vmax: 3394, 3271, 2931, 1775, 1627, 1606, 1594, 1513, 1458, 1376, 1350, 1333, 1319, 1265, 1205, 1174, 1147, 1124, 1063, 1021, 996, 960, 882, 847, 824, 792 cm−1; 1H and 13C NMR data assigned in Table 1; HMBC and NOESY correlations, see Fig. 2; HRESIMSm/z 485.1230 [M+H]+ (calcd. for [C28H21O8]+, 485.1231), 502.1495 (calcd. for [C28H20O8NH4]+, 502.1496), 507.1047 (calcd. for [C28H20O8Na]+, 507.1050).
Methyl etherization of compound 1.
Vaticahainol A (1, 10 mg) was allowed to react with K2CO3 (500 mg) and MeI (200 mg) in dry acetone under reflux for 3 h. Upon completion of the reaction, the resulted mixture was dried through N2 stream, dissolved in CHCl3 (10 ml), washed successively with water (15 ml) and brine (15 ml), and then dehydrated with MgSO4, followed by in vacuoevaporation of the organic solvent. The subsequent gel filtration of the resultant solid over Sephadex LH-20 column with CHCl3/MeOH (1:1) gave the anticipated methyl ether 1a (11 mg, 90% yield) as a white amorphous powder; 1H NMR (500 MHz, acetone-d6): 7.74 (d, J = 2.4 Hz, H-14b), 7.37 (d, J = 2.4 Hz, H-10a), 6.78 (d, J = 2.4 Hz, H-12a), 6.77 (d, J = 2.4 Hz, H-12b), 6.70 (d, J = 8.5 Hz, H-2a, 6a), 6.27 (brs, 4H, H-2b, 3b, 5b, 6b), 6.18 (d, J = 8.5 Hz, H-3a,5a), 5.54 (d, J = 4.2 Hz, H-7a), 4.95 (d, J = 4.2 Hz, H-8a), 3.92 (s, OMe), 3.89 (s, OMe), 3.88 (s, OMe), 3.61 (s, OMe), 3.58 ppm (s, OMe). HRESIMS: m/z: found 555.2010, calcd. for C33H31O8, 555.2013 [M+H]+.
R- and S-MTPA esterification of compound 1.
Duplicate dry samples (each 1 mg, 1.8 × 10−6 mol, 1.0 equiv.) of 1a were dissolved in 0.5 mL of newly distilled dry pyridine and stirred for 20 min. 10 μL of R- and S-MTPA chloride (2.9 × 10−5 mol, 16.1 equiv.) were added respectively to the separate reaction vials and the reaction mixtures were stirred at room temperature for 3 h. The reactions were monitored by LC/MS. After 1 h, MTPA-chloride was quenched by adding 100 μL of methanol. The acylation products (1b and 1c) were purified by reversed-phase HPLC (Hitachi HPLC 7100 series, Waters C18 semi-preparative column, 10 mm × 250 mm, 2 mL/min, MeOH/H2O: 87/13). S- (1b) and R-MTPA esters (1c) were eluted at the retention time of 13.4 and 14.1 min, respectively. The molecular formulas for 1b and 1c were confirmed as C43H37F3O10 by ESI-LC/MS analysis ([M+H]+m/z at 771.2). The 1H chemical shifts around the stereogenic center of 1b and 1c were assigned unequivocally by 1H NMR, COSY and NOESY analyses.
S-MTPA ester (1b).
1H NMR (500 MHz, CDCl3) δ = 7.74 (d, J = 2.0 Hz, H-12b), 6.69 (s, H-14b), 6.68(s, H-12a), 6.59(d, J = 8.0 Hz, H-2a,6a), 6.51(s, H-10a) ,6.36(d, J = 4.5 Hz, H-8a), 6.19(brs, H-2b,3b,5b,6b), 6.13(d, J = 8.0 Hz,H-3a,5a), 5.95 (d, J = 4.5 Hz, H-7a), 3.94 (s, OMe-11b), 3.91 (s, OMe-13b), 3.61 (s, OMe-4a), 3.60 (s, OMe-11a), 3.59 ppm (s, OMe-4b).
R-MTPA ester (1c).
1H NMR (500 MHz, CDCl3) δ = 7.77 (d, J = 2.0 Hz, H-12b), 6.82 (s, H-14b), 6.72(s, H-12a), 6.67(s, H-10a), 6.49(d, J = 8.0 Hz, H-2a,6a), 6.35(d, J = 4.5 Hz, H-8a), 6.20(brs, H-2b,3b,5b,6b), 6.06(d, J = 8.0 Hz,H-3a,5a), 5.90(d, J = 4.5 Hz, H-7a), 3.93 (s, OMe-13b), 3.92 (s, OMe-11b), 3.82 (s, OMe-11a), 3.60 (s, OMe-4b), 3.57 ppm (s, OMe-4a).
Vaticahainol B (2).
Yellow amorphous powder; UV (MeOH) λmax (log ε): 272 (3.8), 312 (3.6), 376 (4.0) nm; IR (KBr) vmax: 3175, 1773, 1647, 1598, 1535, 1512, 1440, 1356, 1317, 1209, 1173, 1108, 1076, 1015, 996, 830 cm−1; 1H and 13C NMR data assigned in Table 1; HMBC and NOESY correlations, see Fig. 5; HRESIMSm/z 485.1232 [M+H]+ (calcd. for [C28H21O8]+, 485.1231), 507.1049 (calcd. for [C28H20O8Na]+, 507.1050)
Vaticahainol C (3).
Yellow amorphous powder; UV (MeOH) λmax (log ε): 248 (4.3), 268 (4.4), 328 (4.0) nm; IR (KBr) vmax: 3208, 2926, 2854, 1612, 1538, 1511, 1448, 1361, 1224, 1175, 1125, 1070, 1052, 1025, 1004, 984, 872, 823 cm−1; 1H and 13C NMR data assigned in Table 1; HMBC and ROESY correlations, see Fig. 7; HRESIMSm/z 467.1124 [M+H]+ (calcd. for [C28H19O7]+, 467.1125), 489.0943 (calcd. for [C28H18O7Na]+, 489.0945).
XO inhibition assay
The XO inhibitory activity was assayed by a described method with some modification.28Xanthine (3.0 mg) was dissolved in 100 ml of 0.1 M phosphate buffer (pH 7.8) to make 200 μM of xanthine buffer. The test solution was prepared by dissolving samples in DMSO, and the assay mixture consisted of 10 μl of test solution and 990 μl of xanthine buffer was prepared for the reaction. The reaction was initiated by the addition of 2 μl enzyme solution (20 units/ml). The mixture was vortexed and incubated at RT for 4 min. The concentration of uric acid was measured by optical density (OD) at 295 nm. Allopurinol was used as standard sample. The inhibitory percentage of xathine oxidase was calculated as follows:
| % Inhibition= [(A–B) − (C–D)]/(A–B) × 100% |
Where A is the OD at 295 nm with enzyme, but without sample; B is the OD at 295 nm without enzyme or sample; C is the OD at 295 nm with enzyme and sample; and D is the OD at 295 nm with sample, but without enzyme.
AChE inhibition assay
The AChE inhibitory activities were measured by a spectrophotometric method developed by Ellman.29 The assay solution was consisted of 130 μl of 0.1 M phosphate buffer (pH 8.0), 20 μl of 0.333 mM DTNB, and 20 μl of 0.035 unit/ml AChE divided from electric eel (480 u/ml, Sigma). 10 μl of test compound solution was then added to the assay solution and pre-incubated for 20 min, followed by the addition of 20 μl of 0.53 mM ATCI. The reaction was measured at 412 nm (37 °C) after 15 min. Five different concentrations of each compound were used in order to obtain the inhibition of AChE activity. The inhibition of AChE activity is expressed as the concentration that results in half-maximal enzyme (IC50).
Acknowledgements
This work was co-supported by grants for from Natural Science Foundation of China (20802035 and 30821006) and Ministry of Education (200802841022).
References
- T. Shen, X. N. Wang and H. X. Lou, Nat. Prod. Rep., 2009, 26, 916–935 RSC.
- H. M. Ge, C. Xu, X. T. Wang, B. Huang and R. X. Tan, Eur. J. Org. Chem., 2006, 5551–5554 CrossRef.
- M. Ohyama, T. Tanaka, T. Ito, M. Iinuma, K. F. Bastow and K. Lee, Bioorg. Med. Chem. Lett., 1999, 9, 3057–3060 CrossRef CAS.
- H. M. Ge, W. H. Yang, J. Zhang and R. X. Tan, J. Agric. Food Chem., 2009, 57, 5756–5761 CrossRef CAS.
- J. R. Dai, Y. F. Hallock, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1998, 61, 351–353 CrossRef CAS.
- M. U. S. Sultanbawa, S. Surendrakumar and P. Bladon, J. Chem. Soc., Chem. Commun., 1980, 619–620 RSC.
- H. M. Ge, W. H. Yang, Y. Shen, N. Jiang, Z. K. Guo, Q. Luo, Q. Xu, J. Ma and R. X. Tan, Chem.–Eur. J., 2010, 6, 6338–6345 CrossRef.
- H. M. Ge, C. H. Zhu, D. H. Shi, L. D. Zhang, D. Q. Xie, J. Yang, S. W. Ng and R. X. Tan, Chem.–Eur. J., 2008, 14, 376–381 CrossRef CAS.
- J. Y. Liu, Y. H. Ye, L. Wang, D. H. Shi and R. X. Tan, Helv. Chim. Acta, 2005, 88, 2910–2917 CrossRef CAS.
- T. Tanaka, T. Ito, K. Nakaya, M. Iinuma and S. Riswan, Phytochemistry, 2000, 54, 63–69 CrossRef CAS.
- T. Ito, T. Tanaka, Y. Ido, K. Nakaya, M. Iinuma, Y. Takahashi, H. Naganawa and S. Riswan, Heterocycles, 2001, 55, 557–567 CrossRef CAS.
- T. Ito, T. Tanaka, M. Iinuma, I. Iliya, K. Nakaya, Z. Ali, Y. Takahashi, R. Sawa, Y. Shirataki, J. Murata and D. Darnaedi, Tetrahedron, 2003, 59, 5347–5363 CrossRef CAS.
- E. K. Seo, H. Chai, H. L. Constant, T. Santisuk, V. Reutrakul, C. W. W. Beecher, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto and A. D. Kinghorn, J. Org. Chem., 1999, 64, 6976–6983 CrossRef CAS.
- Sahidin, E. H. Hakim, L. D. Juliawaty, Y. M. Syah, L. Din, E. L. Ghisalberti, J. Latip, I. M. Said and S. A. Achmad, Z. Naturforsch., C: Biosci., 2005, 60, 723–727 CAS.
- T. Tanaka, T. Ito, Y. Idoa, T. K. Son, K. Nakaya, M. Iinuma, M. Ohyama and V. Chelladuraic, Phytochemistry, 2000, 53, 1015–1019 CrossRef CAS.
- Y. H. He, K. Terashima, Y. Takaya and M. Niwa, Chin. J. Nat. Med., 2005, 3, 90–92 CAS.
- Y. Oshima, Y. Ueno, K. Hisamichi and M. Takeshita, Tetrahedron, 1993, 49, 5801–5804 CrossRef CAS.
- H. F. Luo, L. P. Zhang and C. Q. Hu, Tetrahedron, 2001, 57, 4849–4854 CrossRef CAS.
- T. Tanaka, T. Ito, Y. Ido, K. Nakaya, M. Iinuma and V. Chelladurai, Chem. Pharm. Bull., 2001, 49, 785–787 CrossRef CAS.
- Y. Takaya, K. X. Yan, K. Terashima, J. Ito and M. Niwa, Tetrahedron, 2002, 58, 7259–7265 CrossRef CAS.
- T. Tanaka, T. Ito, K. Nakaya, M. Iinuma, Y. Takahashi, H. Naganawab and S. Riswan, Heterocycles, 2001, 55, 729–740 CrossRef CAS.
- J. R. Zgoda-Pols, A. J. Freyer, L. B. Killme and J. R. Porter, J. Nat. Prod., 2002, 65, 1554–1559 CrossRef CAS.
- M. U. S. Sultanbawa, S. Surendrakumar, M. I. M. Wazeer and P. Bladon, J. Chem. Soc., Chem. Commun., 1981, 23, 1024–1026 Search PubMed.
- W. W. Li, B. G. Li and Y. Z. Chen, Chin. Chem. Lett., 1998, 9, 735–736 CAS.
-
(a) S. He, B. Wu, Y. J. Pan and L. Y. Jiang, J. Org. Chem., 2008, 73, 5233–5241 CrossRef CAS;
(b) K. S. Huang, M. Lin and G. F. Cheng, Phytochemistry, 2001, 58, 357–362 CrossRef CAS.
- J. X. Zhu, Y. Wang, L. D. Kong, C. Yang and X. Zhang, J. Ethnopharmacol., 2004, 93, 133–140 CrossRef CAS.
- S. F. Tsai and S. S. Lee, J. Nat. Prod., 2010, 73, 1632–1635 CrossRef CAS.
- P. Cos, L. Ying, M. Calomme, J. P. Hu, K. Cimanga, B. Van Poel, L. Pieters, A. J. Vlietinck and D. Vanden Berghe, J. Nat. Prod., 1998, 61, 71–76 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. Andres Jr. and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.