One-pot synthesis and characterization of well defined core–shell structure of FePt@CdSe nanoparticles†
Thuy T.
Trinh
a,
Derrick
Mott
a,
Nguyen T. K.
Thanh
bc and
Shinya
Maenosono
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology (JAIST), 1-1 Asahidai, Nomi, 923-1292, Japan. E-mail: shinya@jaist.ac.jp (S. M.); Fax: +81-761-51-1625; Tel: +81-761-51-1611
bThe Davy-Faraday Research Laboratory, The Royal Institution of Great Britain, 21 Albemarle Street, London, W1S 4BS, UK
cDepartment of Physics & Astronomy, University College London, Gower Street, London, WC1E 6BT, UK
First published on 25th July 2011
Abstract
Magnetic fluorescent FePt@CdSe core–shell nanoparticles were directly synthesized by sequential addition of precursors and using tetraethylene glycol as a solvent and a reducing agent. The core–shell NPs were successfully formed over a wide range of temperature (240–300 °C). The size and composition of the FePt core were tuned by changing the ratio of surfactant (oleic acid and oleylamine) to metal precursors [Fe3(CO)12 and Pt(acac)2] and the feeding ratio of the precursors, respectively. The CdSe shell thickness also could be varied from 1 to 8.5 nm by rational control of the total amount of Cd and Se precursors. FePt@CdSe core–shell NPs with a core size of about 4.3 nm and shell thickness of about 2.5 nm displayed a fluorescence emission around 600 nm. They exhibited superparamagnetic behaviour at room temperature and the blocking temperature was about 55 K, which was almost the same as uncoated FePt NPs, while the coercivity decreased from 400 Oe for the FePt NPs to 200 Oe. Detailed characterization of intermediates and synthesized FePt@CdSe NPs revealed the fine structure and formation mechanism.
Introduction
Magnetic-fluorescent hybrid materials composed of magnetic nanoparticles (MNPs) and semiconductor quantum dots (QDs) in novel heteronanostructures have received much attention because they promisingly open up a new window for bioapplications.1 These materials can exhibit properties of the different components in the hybrid structure. The properties of each component can be modified by tuning the conjugate. MNPs show many advantages in bioapplications due to their unique ability to respond to an external magnetic field, which has led to successful applications including protein separation and drug delivery. QDs as fluorescent probes have found increased applications for cell labeling, tracking of cell migration and in vivo imaging. The combination of superparamagnetism and fluorescence at the nanometre scale could lead to new and effective applications in biological systems.2–6In general, hybrid nanoparticles (NPs) can be synthesized either by a direct synthesis without any separation process of a first component,7–9 or by a seed-mediated growth of a second component on pre-synthesized NPs.10–23 In the latter case, the key is controlling heteronucleation/growth of the second component on the seeds in an orderly fashion, and obviously it is not an easy task. The synthesized magnetic-fluorescent hybrid NPs can be classified according to morphology, such as core–shell,9–15 hetero-dumbbell, dimers or trimer,1,7,9,15–19 and sponge or rod-like heterostuctures.8,9,20–24 Among them, the isotropic core–shell NPs are advantageous in terms of biomedical applications, because the NP surface is uniform, and thus, its properties can be tailored and controlled.6 However, the synthesis of the magnetic-fluorescent core–shell NPs is the most difficult probably because there is usually a large lattice mismatch between magnetic core and semiconductor shell.
Few attempts have been made to synthesize MNP@QD core–shell NPs. In most synthetic approaches previously reported, a multistep procedure was employed to obtain core–shell NPs, where MNPs were synthesized and then separated from a reaction solution, followed by purification and the crystal growth of semiconductor shell. There are few reports regarding a one-pot synthesis of the MNP@QD core–shell NPs. One of the few studies is reported by Gao and coworkers.9 As reported, FePt@CdX (X: Se, S) core–shell nanostructures were synthesized via the sequential addition of Cd and then Se (or S) precursors without any separation of FePt NPs in the presence of nonpolar solvents such as phenyl ether, benzyl ether or octyl ether. In their synthetic approach, the FePt@CdSe core–shell NPs were formed by adding Cd precursor [cadmium(II) acetylacetonate] and Se powder sequentially to a reaction mixture containing pre-formed FePt MNPs under a low reaction temperature (ca. 256 °C) in a glove box. When the reaction temperature was increased, the morphology of the synthesized core–shell NPs resulted in heterodimer structures. This means that the experimentally variable temperature range is quite narrow to obtain uniform FePt@CdSe NPs. In addition, the CdSe shell thickness did not change in the experiments and was ∼3.5 nm. It is extremely important to expand the allowable reaction temperature range (wide operability), to make the reaction conditions facile and robust, and to have controllability of shell thickness (emission tunability) from the standpoint of applications. Furthermore, it is also important to study the effect of the CdSe shell on the magnetic properties of the FePt core and to clearly understand the formation mechanism of the CdSe shell, because these issues have not been previously investigated in detail.
In this research, we developed an efficient and facile chemical synthesis route towards uniform FePt@CdSe core–shell NPs. Our one-pot synthetic strategy is rather simple and robust compared to previously reported methods. In our synthesis, monodisperse FePt@CdSe core–shell NPs could be synthesized over a relatively wide shell growth temperature range (240–300 °C) without formation of heterodimers. Importantly, the FePt core size and the CdSe shell thickness can be independently varied to some extent. In addition, we closely tracked the formation process of the CdSe shell and clarified the effect of the CdSe shell on the magnetic properties of FePt cores using a wide range of analytical methods including X-ray photoelectron spectroscopy, aberration-corrected scanning transmission electron microscopy, superconducting quantum interference device magnetometry, and many others.
Experimental section
Chemicals
Platinum(II) acetylacetonate [Pt(acac)2, purity 97%], triiron dodecacarbonyl [Fe3(CO)12, purity 99.999%], selenium powder (Se, purity 99.5%), cadmium acetate [Cd(OAc)2, purity 99.99%], tetraethylene glycol (TEG, purity 99%), oleic acid (OA, 99%), oleylamine (OLA, 70%), and trioctylphosphine (TOP, 90%) were purchased from Sigma Aldrich Corp. All reagents were used without further purification.Synthesis of FePt@CdSe core–shell NPs
FePt NPs were synthesized via the thermal decomposition of Fe3(CO)12 (85.37 mg, 0.169 mmol) and the reduction of Pt(acac)2 (100 mg, 0.254 mmol) in TEG (20 mL) in the presence of OA (1 mL) and OLA (1 mL) at T1 = 240 °C for 2 h under an Ar atmosphere, where T1 denotes the temperature for FePt NPs synthesis. After the reaction, the solution was cooled down to 200 °C, and then, the cadmium stock solution was prepared by dissolving Cd(OAc)2 (117.21 mg, 0.508 mmol) in 3.0 mL of TOP, which was then added into the reaction solution. The reaction temperature was kept at T2 = 200 °C for 30 min under an Ar atmosphere to decompose the Cd(OAc)2 precursor to form FePt@CdOx intermediate core–shell nanostructures, where T2 denotes the temperature for CdOx shell growth. The solution color gradually changed from black to dark brown, indicating the formation of CdOx after the thermal decomposition of Cd(OAc)2. After that, the reaction temperature was increased up to T3, and then, the Se stock solution (44.17 mg, 0.559 mmol of Se powder dissolved in 0.6 mL TOP) was quickly injected into the reaction mixture under vigorous stirring, where T3 denotes the temperature for selenization of the CdOx shell. T3 was varied to 240, 260, and 300 °C in the present study. The injection of the Se-TOP complex into the reaction mixture promotes a selenization of the CdOx shell. After 30 min of reaction at T3, the flask was cooled to room temperature and ethanol/hexane mixture was added to the flask. By centrifuging this mixture, a black powder was separated from the matrix. The powder was then redispersed in a solution of 0.5 vol % OLA in hexane.Material characterization and analysis conditions
Synthesized NPs were fully characterized by transmission electron microscopy (TEM), high-resolution TEM (HRTEM), scanning TEM (STEM), energy-dispersive X-ray spectroscopy (EDS), X-ray diffractometry (XRD), X-ray photoelectron spectroscopy (XPS), UV/Vis spectroscopy, fluorescence spectroscopy and superconducting quantum interference device (SQUID) magnetometer. TEM analysis was performed on a Hitachi H-7650 transmission electron microscope operated at 100 kV. HRTEM and EDS analyses were performed on a Hitachi H-9000NAR transmission electron microscope operated at 300 kV equipped with an EDS detector. The core–shell structure was further confirmed by using a JEOL JEM-ARM200F scanning transmission electron microscope, which incorporates a spherical aberration corrector, with an EDS detector. TEM samples were prepared by dropping the NP dispersion onto a carbon coated copper grid and drying in air. XRD patterns were collected in reflection geometry using a Rigaku RINT2500 X-ray diffractometer at room temperature with Cu Kα radiation (wavelength 1.542 Å). XPS analysis was carried out on a Shimadzu Kratos AXIS-ULTRA DLD high performance XPS system. Photoelectrons were excited by monochromated Al Kα radiation. Detection was done with a delay-line detector (DLD) and a concentric hemispherical analyzer (CHA). The X-ray tube was operated at 150 W. The pass energy of the CHA was 20 eV for narrow-scan spectra. The analyzed area on the specimen surface was 300 × 700 μm2 and was located in the center of the irradiated region. For the sample preparation, the dried NP powder was deposited on carbon tape in air. The instrument was operated at a vacuum level of 1 × 10−8 Torr. Magnetic properties of FePt and FePt@CdSe NPs were analyzed by a Quantum Design MPMS SQUID magnetometer. Absorption and fluorescence spectra were recorded on a Perkin-Elmer Lambda 35 UV/Vis spectrometer and a Jasco FP-6300 spectrofluorimeter, respectively.Results and discussion
Formation of FePt MNPs
Scheme 1 illustrates the typical synthetic route towards FePt@CdSe core–shell NPs. By modifying the procedure reported by Jeyadevan et al.25 and Kang et al.,26 the thermal decomposition of Fe3(CO)12 and the reduction of Pt(acac)2 at 240 °C in TEG as solvent and reducing agent and in the presence of surfactants including OA and OLA produces FePt MNPs (A).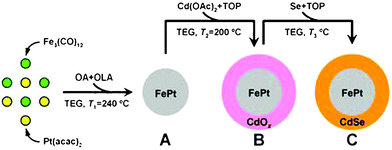 | ||
| Scheme 1 Synthetic approach towards FePt@CdSe core–shell NPs. | ||
Size, size distribution, shape and crystalline structure of FePt MNPs synthesized were analyzed by TEM images and XRD patterns as shown in Fig. 1. By increasing the concentration of the surfactants while keeping the OA:OLA volume ratio constant (= 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the mean size (Dcore) could be tuned in the range 3–5 nm, i.e. 3.2 nm (OA + OLA = 3.1 mmol, Fig. 1a), 4.2 nm (6.2 mmol, Fig. 1b) and 4.7 nm (9.3 mmol, Fig. 1c). The crystal structure of FePt MNPs was chemically-disordered face-centered cubic (fcc) phase. The mean crystallite sizes (Dxrd) estimated from the full width at half-maximum of the (111) peak by the Scherrer formula were 2.2, 2.8 and 2.8 nm for Fig. 1a, b and c, respectively. Compositions of these MNPs estimated by EDS were Fe61Pt39, Fe64Pt36 and Fe50Pt50, respectively. FePt MNPs have a spherical shape with a narrow size distribution (CV < 6%). The uniformity of FePt MNPs in this study is improved over those previously reported for FePt MNPs synthesized in TEG,25 probably due to the presence of OA and OLA as stabilizers. In fact, the as-synthesized FePt MNPs could easily self-assemble into an ordered superlattice (Fig. S1, ESI†).
:1), the mean size (Dcore) could be tuned in the range 3–5 nm, i.e. 3.2 nm (OA + OLA = 3.1 mmol, Fig. 1a), 4.2 nm (6.2 mmol, Fig. 1b) and 4.7 nm (9.3 mmol, Fig. 1c). The crystal structure of FePt MNPs was chemically-disordered face-centered cubic (fcc) phase. The mean crystallite sizes (Dxrd) estimated from the full width at half-maximum of the (111) peak by the Scherrer formula were 2.2, 2.8 and 2.8 nm for Fig. 1a, b and c, respectively. Compositions of these MNPs estimated by EDS were Fe61Pt39, Fe64Pt36 and Fe50Pt50, respectively. FePt MNPs have a spherical shape with a narrow size distribution (CV < 6%). The uniformity of FePt MNPs in this study is improved over those previously reported for FePt MNPs synthesized in TEG,25 probably due to the presence of OA and OLA as stabilizers. In fact, the as-synthesized FePt MNPs could easily self-assemble into an ordered superlattice (Fig. S1, ESI†).
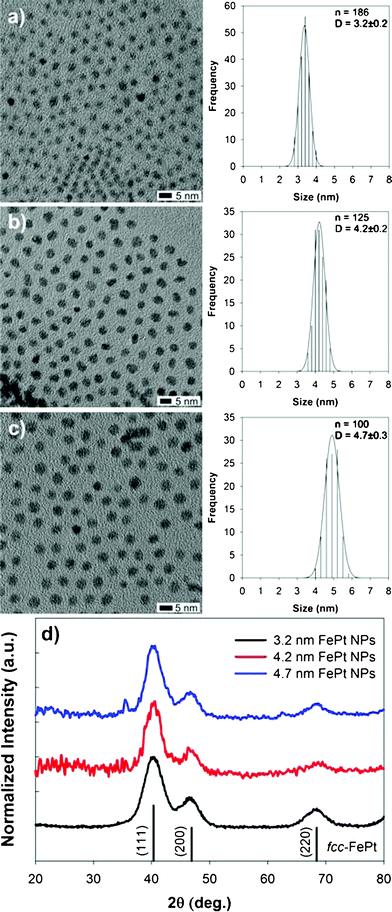 | ||
| Fig. 1 TEM images and size distributions of FePt MNPs synthesized with OA + OLA = 3.1 mmol (Dcore = 3.2 nm) (a), 6.2 mmol (4.2 nm) (b) and 9.3 mmol (4.7 nm) (c), and XRD patterns of each sample (d). | ||
The Fe content of FePt MNPs linearly increased with increasing the amount of Fe precursor (data not shown). The enhanced composition controllability compared to similar syntheses in which iron pentacarbonyl [Fe(CO)5] is used as Fe precursor can be understood because Fe3(CO)12 does not vaporize during the reaction while Fe(CO)5 easily vaporizes and thus produces inhomogeneity in the reaction mixture. Note that the decomposition temperature of Fe3(CO)12 is about 90 °C (Fig. S2, ESI†).
Formation of intermediate core–shell NPs
In the present synthetic procedure, the Cd precursor [Cd(OAc)2] was injected to form intermediate core–shell NPs (B) without separation or purification of the FePt MNPs as explained in the Experimental Section and shown in Scheme 1. The reaction between hydroxyl groups in TEG and acetate resulted in the formation of the corresponding ester.10 Sequential addition of Cd(OAc)2 into the reaction mixture is an important process to control the final structure. In general, Cd was deposited onto the core to form a core–shell structure, eventually leading to the formation of a FePt@CdSe core–shell structure after the injection of Se precursor.7,9,10,15 For instance, the FePt@CdO core–shell structure might be easily formed because CdO can be smoothly grown onto FePt MNP surfaces in the form of both amorphous and crystal phase, which has the same fcc structure as FePt, as mentioned by Gao et al.9 However, there was little experimental evidence indicating the existence of CdO shell intermediate in the past literature.Fig. 2a shows a TEM image of intermediate NPs. In Fig. 2a, we could see only FePt cores. Fig. 2b shows the XRD pattern of intermediate NPs. As seen in Fig. 2b, the main peaks are coming from the fccFePt phase and several minor peaks are from iron oxide phase which is presumably due to the surface oxidation of MNPs. No peaks corresponding to Cd or CdO were observed in the XRD pattern. Based on TEM and XRD results, there was no indication of CdO shell formation on FePt cores. Therefore, we conducted XPS analysis to investigate whether a CdO shell is formed on FePt cores.
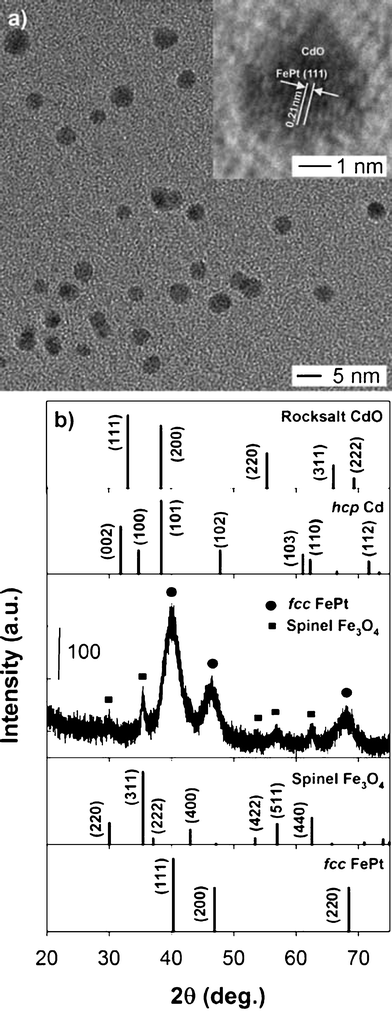 | ||
| Fig. 2 (a) TEM image of intermediate NPs (B). The inset shows a HRTEM image of a single NP. (b) XRD pattern of intermediate NPs with reference peaks for fccFePt (JCPDS card no. 00-029-0718), Fe3O4 (01-079-0419), Cd (00-005-0674) and CdO (003-065-2908). Composition measured by EDS is Fe:Pt:Cd = 37:38:25. | ||
Fig. 3a and S3a (ESI†) show high resolution XPS core-level spectra of FePt MNPs (A). The Fe 2p core levels are split into 2p1/2 and 2p3/2 components, which is due to spin–orbit coupling. To take a closer look at the high resolution Fe 2p spectra, the Fe 2p3/2 component, which is asymmetrically broadened due to an existence of high-spin Fe cation and/or symmetry breaking at the surface, was deconvoluted by using four Gaussian functions corresponding to Fe(0) component and Gupta and Sen (GS) triplets.27 Full width at half maximum (FWHM), relative intensity and peak energy of each deconvoluted Gaussian peak were within ranges broadly consistent with previously reported values.27,28
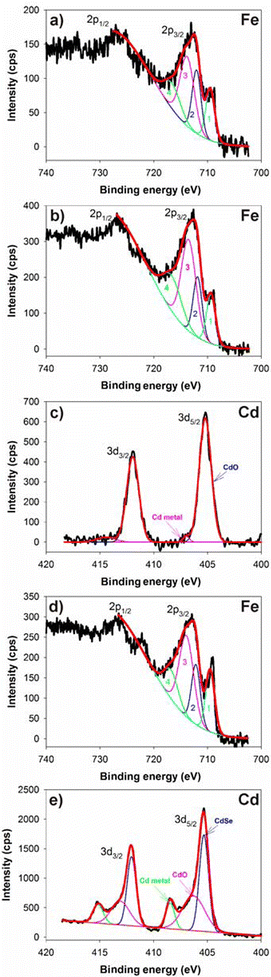 | ||
| Fig. 3 XPS spectra of FePt (A), FePt@CdOx (B) and FePt@CdSe260 (C) NPs. (a), (b) and (d) are Fe 2p spectra for A, B and C, respectively. (c) and (e) are Cd 3d spectra for B and C, respectively. | ||
As shown in Fig. 3a, the Fe 2p3/2 line shape was divided into four peaks: first is at a binding energy of 709.5 eV (Peak 1), second is at 712.1 eV (Peak 2), third is at 713.9 eV (Peak 3), and forth is at 717.5 eV (Peak 4). The Peaks 2, 3 and 4 are GS triplets and correspond to an oxidized state of Fe, while the Peak 1 corresponds to Fe(0).28,29 The slight shift in binding energy of Fe 2p3/2 compared to reference data of bulk standard is presumably due to initial and final state effects of the electron emission process in small particles.30 The Pt 4f peaks were also split into two spin–orbit doublets 4f7/2 and 4f5/2 as shown in Fig. S3a (ESI†). The Pt 4f7/2 peak can be divided into two peaks: one is at a binding energy of 71.0 eV and the other is at 71.7 eV. The Pt 4f5/2 peak also can be divided into two peaks: one is at a binding energy of 74.5 eV and the other is at 75.2 eV. The low energy peaks observed in both 4f7/2 and 4f5/2 components are indicative of Pt(0), whereas the high energy peaks were added in order to fit the experimental spectrum correctly.31 The compositions estimated from XPS spectra reasonably agree with those estimated from EDS analyses. For example, the composition of the FePt MNPs shown in Fig. 1b was estimated to be Fe64Pt36 and Fe77Pt23 by EDS and XPS, respectively.
Fig. 3b and S3b (ESI†) show Fe 2p and Pt 4f spectra, respectively, for intermediate NPs (B). The Fe 2p3/2 line shape was divided into four peaks as discussed above. Peaks 1, 2, 3, and 4 are at binding energies of 709.4, 711.9, 713.6, and 717.5 eV, respectively. Fig. 3c shows the Cd 3d spectrum for intermediate NPs (B). The Cd 3d core levels are split into 3d5/2 and 3d3/2 components due to spin–orbit coupling. The Cd 3d5/2 line shape can be divided into two peaks: one intense peak is at a binding energy of 405.6 eV, which corresponds to CdO and/or surface Cd(OH)2. The Cd 3d3/2 line shape can also be divided into two peaks: one intense peak is at a binding energy of 411.9 eV and the other very weak peak is at a binding energy of 414.4 eV. The weak peaks observed in both 3d5/2 and 3d3/2 components are indicative of Cd(0). As can be seen in Fig. S3c (ESI†), no Se peak is observed. The broad peak in Fig. S3c comes from contribution of Fe 3p or Pt 5p with normal distribution centered about 53 and 52 eV, respectively. Considering all the results from TEM, XRD, EDS and XPS analyses, we conclude that the intermediate NPs have an FePt@CdOx core–shell structure and the CdOx shell is amorphous.
Formation of FePt@CdSe core–shell NPs
FePt@CdSe core–shell NPs were formed via subsequent selenization of FePt@CdOx NPs. Fig. 4a, b and c show TEM images of the FePt@CdSe core–shell NPs (C) synthesized at T3 = 240, 260 and 300 °C, respectively. We call these NPs FePt@CdSe240, FePt@CdSe260 and FePt@CdSe300, hereafter. As seen in Fig. 4, FePt@CdSe240 and FePt@CdSe260 are highly spherical and monodispersed core–shell NPs, while FePt@CdSe300 seems to be sintered or ripened. Fig. 4d shows a HRTEM image of FePt@CdSe260 indicating FePt cores are individually surrounded by CdSe crystals. Curves (i), (ii) and (iii) in Fig. 4e show the XRD patterns of FePt NPs, CdSe NPs synthesized at the same reaction conditions and FePt@CdSe260, respectively. It can be clearly observed that FePt@CdSe260 has two phases: one is fccFePt and the other is Wurtzite CdSe.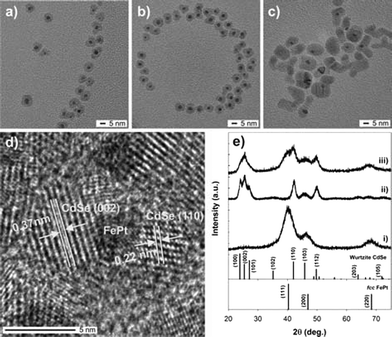 | ||
| Fig. 4 TEM images of the FePt@CdSe core–shell NPs synthesized at (a) T3 = 240, (b) 260 and (c) 300 °C. (d) HRTEM image of FePt@CdSe260. (e) XRD patterns of (i) FePt NPs, (ii) CdSe NPs synthesized at the same reaction conditions and (iii) FePt@CdSe260 NPs with reference peaks for fccFePt (JCPDS card no. 00-029-0718) and CdSe (01-075-5681). | ||
Fig. 3d and S3d (ESI†) show Fe 2p and Pt 4f spectra, respectively, for FePt@CdSe260 NPs (C). The Fe 2p3/2 line shape was divided into four peaks as discussed above. Peaks 1, 2, 3, and 4 are at binding energies of 709.5, 712.2, 714.0, and 717.3 eV, respectively. This result is almost the same as the case of FePt@CdOx NPs. In addition, the intensity of the first peak of the neutral Fe state of Fe 2p3/2 component is relatively more intense for FePt@CdSe260 NPs than for FePt NPs as seen in Fig. 3d and a. This result suggests that the oxidized Fe states in the uncoated FePt MNPs could be caused by the capping ligands that are chemisorbed on Fe sites,32 and the enhanced intensity of the neutral zero valent Fe state in the FePt@CdSe NPs would be observed because the capping ligands were desorbed and replaced with the CdSe shell. Fig. 3e and S3e (ESI†) show Cd 3d and Se 3d spectra respectively for FePt@CdSe260 NPs. As shown in Fig. 3e, the Cd 3d5/2 line shape can be divided into three peaks: the first intense peak is at a binding energy of 405.3 eV, which corresponds to CdSe.33 Second is at a binding energy of 406.3 eV, which corresponds to CdO and a surface composed of Cd(OH)2.33 Third is at a high binding energy of 408.5 eV that can be assigned to Cd0.34 The Cd 3d3/2 line shape can also be divided into three peaks. As can be seen in Fig. S3e, the Se 3d core levels are split into 3d5/2 (54.5 eV) and 3d3/2 (56.9 eV) components.33 An existence of the CdO peak whose amplitude is almost equal to the area of the CdSe peak suggests an incomplete selenization. In addition, the atomic ratio of Cd to Se is always larger than 1. These results suggest that an existence of the amorphous CdO interfacial layer between FePt core and CdSe shell. Note that the atomic ratio of Cd to Se did not significantly vary by decreasing the shell thickness (see Table 1).
| Sample Name | T 3 (°C) | Core | Shell | ||
| D core (nm) | Composition (atomic %) | Thickness (nm) | Composition (atomic %) | ||
| FePt@CdSe240 | 240 | 4.2 ± 0.3 | Fe53Pt47 | 2.4 ± 0.4 | Cd62Se38 |
| FePt@CdSe260 | 260 | 4.3 ± 0.3 | Fe54Pt46 | 2.5 ± 0.4 | Cd57Se43 |
| FePt@CdSe300 | 300 | 4.2 ± 0.5 | Fe51Pt49 | − | Cd64Se36 |
| FePt@CdSe260thin | 260 | 4.3 ± 0.3 | Fe54Pt46 | ∼0.7 | Cd56Se44 |
| FePt@CdSe260thick | 260 | 4.3 ± 0.3 | Fe54Pt46 | ∼8.5 | N/A |
To further confirm the FePt@CdSe core–shell structure, high-angle annular dark-field (HAADF) STEM imaging and two-dimensional EDS elemental mapping of individual NPs were performed. Fig. 5 shows the STEM image of a single NP containing relatively heavy elements at the core and lighter elements in the shell (Z contrast). This result is consistent with the FePt@CdSe core–shell structure. Fig. 6 shows the results of EDS elemental mapping. This is stark evidence of the FePt@CdSe core–shell structure showing that Fe (Fig. 6c) and Pt (Fig. 6d) are localized in a core area in HAADF-STEM, while Cd (Fig. 6f) and Se (Fig. 6e) are localized in a shell configuration.
 | ||
| Fig. 5 HAADF-STEM images of two different single NPs in the FePt@CdSe260 sample. Core size and shell thickness are estimated to be (a) 4.6 nm and 2.3 nm, and (b) 4.1 nm and 2.1 nm, respectively. | ||
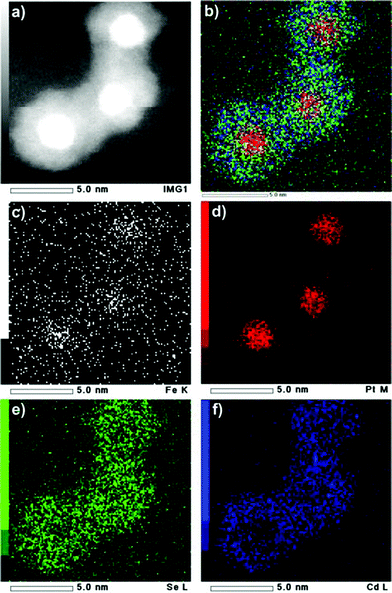 | ||
| Fig. 6 HAADF-STEM image (a) and EDS elemental mapping images of FePt@CdSe260 NPs (b–f): overlay (b) of Fe K edge (c), Pt M edge (d), Se L edge (e), and Cd L edge (f) illustrates the core–shell structure of the FePt@CdSe NPs. | ||
In this section we briefly discuss the FePt@CdSe NP morphology dependence on the shell growth temperature (T3). Table 1 summarizes the results of FePt@CdSe NPs synthesized at three different T3 (240, 260 and 300 °C). It was found that the core–shell structure can be formed under any of these temperatures (Fig. 4). While FePt@CdSe300 (Fig. 4c) showed some degree of fusion of NPs and some fraction of unassociated CdSe NPs due to the homogeneous nucleation and growth of CdSe NPs at high temperature, FePt@CdSe240 (Fig. 4a) and FePt@CdSe260 (Fig. 4b) NPs exhibited much better monodispersity. In the previous study, it has been reported that an FePt-CdSe heterodimer structure was mainly formed in a similar reaction using nonpolar solvents, such as benzyl ether and dioctyl ether at a high temperature (ca. 300 °C).9 This indicates the significant role played by the TEG polar solvent, that is, it can prevent the dewetting of CdSe from FePt cores. Another possibility is that a polar solvent replenishes electrons which are depleted from the cores by the first nucleation of a second component, and then facilitates multi-nucleation sites, eventually forming a continuous shell, even at a high reaction temperature.3 Whatever the case, we could synthesize FePt@CdSe core–shell NPs even at higher temperature (compared to previous studies) by using TEG as the solvent and reducing agent. However, shell thickness and composition could not be controlled at T3. In addition, the FePt core size was unchanged upon the selenization.
As a next step, the total amount of Cd and Se precursors was changed to vary the CdSe shell thickness. By increasing the total amount of the precursors, X = [Cd(OAc)2] + [Se-TOP], keeping Cd:Se molar ratio constant (= 1:1.1), the shell thickness was found to readily increase as shown in Table 1 and Fig. S4 (ESI†). Specifically, the average shell thicknesses were <1 nm (Fig. S4a), 2.5 nm (Fig. 4b and S4b) and 8.5 nm (Fig. S4c) when X = 0.5, 1.1 (standard condition) and 2.1 mmol, respectively. We call the first and third set of NPs FePt@CdSe260thin and FePt@CdSe260thick, hereafter. Because FePt@CdSe260thin NPs showed some degree of fusion of NPs and some fraction of unassociated CdSe NPs possibly due to the dewetting of CdSe from FePt cores (Fig. S4a), we could not determine the shell thickness precisely. Therefore, we estimated the average shell thickness based on the EDS result. The composition of FePt@CdSe260thin was measured as Fe26Pt20Cd30Se24. Assuming a core–shell NP has a concentric spherical shape, the average shell thickness was calculated to be 0.7 nm. Based on these results, it can be emphasized that our synthetic approach has an advantage in the controllability of the CdSe shell thickness.
The selenization process of FePt@CdOx core–shell NPs was further investigated in the reaction at 260 °C. A trace amount of the reaction solution was sampled after the elapse of a certain period of time from the addition of Se stock solution. It was found that 5 min after the addition of the Se stock solution, some core–shell NPs had been produced as shown in Fig. S5a (ESI†). In the case of FePt@CdOx NPs, the shell structure was obscured in the TEM image, probably due to its amorphous nature as shown in Fig. 2a. After the 5 min selenization reaction, however, the shell material becomes easily observable in the TEM image. When the reaction time was prolonged to 10 min, the selenization seems to be almost completed as shown in Fig. S5b. When the reaction time was prolonged to 30 or 60 min, there was almost no variation in the NP morphology, i.e., core size, shell thickness, and size distribution, as shown in Fig. S5c and d. This result indicates that FePt@CdSe core–shell NPs can be quickly formed (within 10 min) in contrast to the previous study in which the shell formation was incomplete even after the 30 min reaction.9
Optical and magnetic properties
Absorption and fluorescence spectra of the as-synthesized FePt@CdSe260 NPs are shown in Fig. 7. The fluorescence intensity is relatively low compared to CdSe NPs, presumably due to the electron transfer from CdSe shell to the FePt metallic core.9 The fluorescence quenching might be prominent when the interface between FePt and CdSe is more coherent. The coherency of the interface is clearly observed in the HAADF-STEM image (Fig. 5), facilitating their interaction. Although the as-synthesized FePt@CdSe NPs have a shell thickness of 2.5 nm, their emission peak is at 604 nm. Gao and coworkers reported that a fluorescence emission peak of FePt@CdSe core–shell NPs with a 10 nm outer diameter and 3 nm FePt core diameter was observed at 465 nm,9 which is similar to that reported for 2 nm CdSe NPs.35 In their case, the shell thickness and volume are estimated to be 3.5 nm and 509.5 nm3, respectively. Note that the CdSe shell volume is almost equal to that of 9.9 nm CdSe NPs. Hence, the quantum confinement effect is expected to be weak. Nonetheless, they observed the blue emission as mentioned before. The observation of the blue emission was attributed to the small size of the CdSe nanoclusters (ca. 2 nm) in shells around the FePt cores. In contrast, for our case, the outer and core diameters of FePt@CdSe260 NPs are 9.3 and 4.3 nm, respectively. Thus, the shell volume is 379.5 nm3, which is almost equal to that of 9 nm CdSe NPs. The emission peak at 604 nm of FePt@CdSe260 NPs is similar to that of 5 nm CdSe NPs.35 This result suggests that the CdSe shell of our core–shell NPs would have better crystallinity compared to that of the previous study.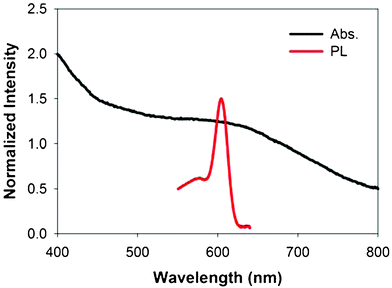 | ||
| Fig. 7 UV/Vis and fluorescence (excitation wavelength = 400 nm) spectra of FePt@CdSe260 NPs. | ||
The blocking temperature (TB) was addressed using zero-field-cooled (ZFC) and field-cooled (FC) measurements. The temperature sweeps were collected under a constant field of 500 Oe. The magnetization (M-H) curve was measured as a function of the applied field at temperatures over TB. Fig. 8 shows temperature dependence of magnetization (FC and ZFC curves) and field dependence of magnetization (M-H curves) at 5 K for 4.2 nm bare Fe64Pt36 NPs and FePt@CdSe260 NPs. The blocking temperatures, estimated from subtraction of ZFC and FC curves, were almost the same for FePt NPs and FePt@CdSe260 NPs, and is approximately 55 K. ZFC curve measured for FePt@CdSe260 NPs exhibited a broad peak as seen in Fig. 8c, suggesting a large anisotropy distribution. The magnetocrystalline anisotropy constant (K) was estimated using the following equation:
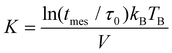 | (1) |
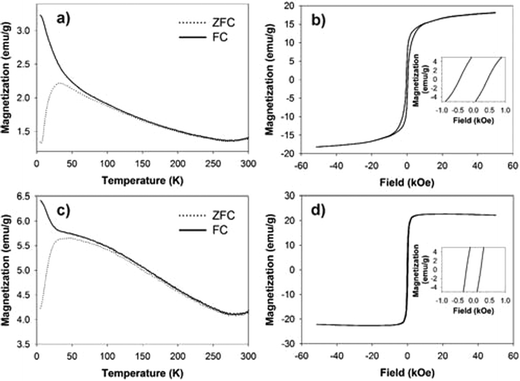 | ||
| Fig. 8 Left: FC and ZFC magnetization versus temperature curves for (a) FePt and (c) FePt@CdSe260 NPs. Right: magnetization versus applied field curves measured at 5 K for (b) FePt and (d) FePt@CdSe260 NPs. The insets in b and d show magnified hysteresis loops for FePt and FePt@CdSe260 NPs. | ||
The formation of FeSe and FeS alloy layers at the interfaces of FePt-PbSe and FePt-PbS systems was observed by Lee et al.14 As reported, the existence of the interfacial layer at the interface between the magnetic core and semiconducting shell caused an increase in TB of these systems compared to those of uncoated FePt. However, in our case, both FePt and FePt@CdSe260 have almost the same TB of 55 K. This might be due to the existence of CdO interfacial layer in between FePt core and CdSe shell suppressing the formation of FeSe interfacial layer.
According to the Stoner–Wohlfarth theory,36 the coercivity of NPs is determined by magnetocrystalline anisotropy constant K and saturation magnetization MS:
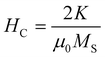 | (2) |
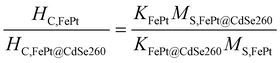 | (3) |
Conclusion
In conclusion, magnetic fluorescent FePt@CdSe core–shell NPs have been directly synthesized over a wide range of temperature (240–300 °C) in polar solventsvia a chemical route. The synthetic method is effective and enables some tuning of FePt core size and composition easily, while CdSe shell thickness proved possible to control by controlling the total amount of Cd and Se precursors. The employment of high resolution XPS and high-angle annular dark-field (HAADF) STEM as well as two-dimensional EDS elemental mapping further revealed the formation mechanism and the structure of core–shell NPs. The materials are highly interesting because FePt@CdSe core–shell NPs revealed both superparamagnetic with enhanced magnetization and fluorescent properties. While the emission efficiency of the material is relatively low, with further study and processing, these materials are promising candidates for biomedical applications.Acknowledgements
The authors thank Dr Koichi Higashimine for his kind help with conducting STEM. Nguyen TK Thanh thanks the Royal Society for her University Research Fellowship. Thuy T. Trinh thanks the Davy Faraday Research Laboratory for hosting his research for a 3 month period.References
- J. Gao, W. Zhang, P. Huang, B. Zhang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2008, 130, 3710 CrossRef CAS.
- A. Quarta, R. D. Corato, L. Manna, A. Ragusa and T. Pellegrino, IEEE Trans. NanoBiosci., 2007, 6, 298 CrossRef.
- H. Zeng and S. Sun, Adv. Funct. Mater., 2008, 18, 391 CrossRef CAS.
- J. Cheon and J. Lee, Acc. Chem. Res., 2008, 41, 1630 CrossRef CAS.
- J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097 CrossRef CAS.
- R. Koole, W. J. M. Mulder, M. M. van Schooneveld, G. J. Strijkers, A. Meijerink and K. Nicolay, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 475 CrossRef CAS.
- H. Gu, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664 CrossRef CAS.
- H. Gu, R. Zheng, H. Liu, X. Zhang and B. Xu, Small, 2005, 1, 402 CrossRef CAS.
- J. Gao, B. Zhang, Y. Gao, Y. Pan, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 11928 CrossRef CAS.
- Z.-Q. Tian, Z.-L. Zhang, P. Jiang, M.-X. Zhang, H.-Y. Xie and D.-W. Pang, Chem. Mater., 2009, 21, 3039 CrossRef CAS.
- X. Liu, Q. Hu, X. Zhang, Z. Fang and Q. Wang, J. Phys. Chem. C, 2008, 112, 12728 CAS.
- M. Zhang, S. Shi, J. Meng, X. Wang, H. Fan, Y. Zhu, X. Wang and Y. Qian, J. Phys. Chem. C, 2008, 112, 2825 CAS.
- H. Kim, M. Achermann, L. P. Balet, J. A. Hollingsworth and V. I. Klimov, J. Am. Chem. Soc., 2005, 127, 544 CrossRef CAS.
- J.-S. Lee, M. I. Bodnarchuk, E. V. Shevchenko and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 6382 CrossRef CAS.
- Z.-Q. Tian, Z.-L. Zhang, J. Gao, B.-H. Huang, H.-Y. Xie, M. Xie, H. D. Abruña and D.-W. Pang, Chem. Commun., 2009, 4025 RSC.
- K.-W. Kwon and M. Shim, J. Am. Chem. Soc., 2005, 127, 10269 CrossRef CAS.
- S. He, H. Zhang, S. Delikanli, Y. Qin, M. T. Swihart and H. Zeng, J. Phys. Chem. C, 2009, 113, 87 CAS.
- K. Tao, H. Zhou, H. Dou, B. Xing, W. Li and K. Sun, J. Phys. Chem. C, 2009, 113, 8762 CAS.
- H. McDaniel and M. Shim, ACS Nano, 2009, 3, 434 CrossRef CAS.
- T. Teranishi, Y. Inoue, M. Nakaya, Y. Oumi and T. Sano, J. Am. Chem. Soc., 2004, 126, 9914 CrossRef CAS.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, M. L. Curri, C. Innocenti, C. Sangregorio, K. Achterhold, F. G. Parak, A. Agostiano and P. D. Cozzoli, J. Am. Chem. Soc., 2006, 128, 16953 CrossRef CAS.
- M. Casavola, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, E. F. Pinel, M. A. Garcia, L. Manna, R. Cingolani and P. D. Cozzoli, Nano Lett., 2007, 7, 1386 CrossRef CAS.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, M. Garcia-Hernandez, M. A. Garcia, R. Cingolani and P. D. Cozzoli, J. Am. Chem. Soc., 2010, 132, 2437 CrossRef CAS.
- J. Maynadié, A. Salant, A. Falqui, M. Respaud, E. Shaviv, U. Banin, K. Soulantica and B. Chaudret, Angew. Chem., Int. Ed., 2009, 48, 1814 CrossRef.
- B. Jeyadevan, K. Urakawa, A. Hobo, N. Chinnasamy, K. Shinoda, K. Tohji, D. D. J. Djayaprawira, M. Tsunoda and M. Takahashi, Jpn. J. Appl. Phys., 2003, 42, L350 CrossRef CAS.
- S. Kang, S. Shi, D. E. Nikles and J. W. Harrell, J. Appl. Phys., 2008, 103, 07D503 CrossRef.
- R. P. Gupta and S. K. Sen, Phys. Rev. B: Solid State, 1975, 12, 15 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564 CrossRef CAS.
- B. Stahl, J. Ellrich, R. Theissmann, M. Ghafari, S. Bhattacharya, H. Hahn, N. S. Gajbhiye, D. Kramer, R. N. Viswanath, J. Weissmüller and H. Gleiter, Phys. Rev. B: Condens. Matter, 2003, 67, 014422 CrossRef.
- G. K. Wertheim, Z. Phys. D: At., Mol. Clusters, 1989, 12, 319 CrossRef CAS.
- C. Dablemont, P. Lang, C. Mangeney, J. -Y. Piquemal, V. Petkov, F. Herbst and G. Viau, Langmuir, 2008, 24, 5832 CrossRef CAS.
- H. G. Bagaria, E. T. Ada, M. Shamsuzzoha, D. E. Nikles and D. T. Johnson, Langmuir, 2006, 22, 7732 CrossRef CAS.
- J. E. B. Katari, V. L. Colvin and A. P. Alivisatos, J. Phys. Chem., 1994, 98, 4109 CrossRef CAS.
- D. Oyetunde, M. Afzaal, M. A. Vincent, I. H. Hillier and P. O’Brien, Inorg. Chem., 2011, 50, 2052 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- E. C. Stoner and E. P. Wohlfarth, Philos. Trans. R. Soc. London, Ser. A, 1948, 240, 599 CrossRef.
- X. W. Wu, C. Liu, L. Li, P. Jones, R. W. Chantrell and D. J. Weller, J. Appl. Phys., 2004, 95, 6810 CrossRef CAS.
- T. Thomson, M. F. Toney, S. Raoux, S. L. Lee, S. Sun, C. B. Murray and B. D. Terris, J. Appl. Phys., 2004, 96, 1197 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: TEM image of superlattice of FePt NPs, TGA result for Fe3(CO)12, TEM images of FePt@CdSe260 NPs formed at different concentration of Cd(OAc)2 and Se precursors and TEM images of FePt@CdSe260 NPs formed in reactions lasting amounts of time. See DOI: 10.1039/c1ra00012h/ |
| This journal is © The Royal Society of Chemistry 2011 |