Controllable Morphology and Photocatalytic Performance of Bismuth Silicate Nanobelts/Nanosheets†
Peiyang
Zhang
,
Juncheng
Hu
* and
Jinlin
Li
Key Laboratory of Catalysis and Materials Science of the State Ethnic Affairs Commission & Ministry of Education, Hubei Province, South-Central University for Nationalities, Wuhan, China. E-mail: junchenghuhu@hotmail.com; Fax: +86 27 67841302; Tel: +86 27 67842752;
First published on 27th September 2011
Abstract
Bismuth silicate (BSO) nanobelts/nanosheets were successfully synthesized for the first time via a one-pot hydrothermal method. The samples were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), high-resolution TEM (HRTEM), N2adsorption-desorption (BET), and UV-vis diffuse reflectance spectroscopy (DRS). The results showed that the concentration of cetyltrimethyl ammonium bromide (CTAB) had crucial effect on the formation of the phase and structure of BSO nanobelts/nanosheets, CTAB acted as a structure-directing reagent. A morphology-controlled process and formation mechanism are proposed. The bismuth silicate samples showed higher photocatalytic activity for the degradation of phenol and azo dye RhB than the commercial catalyst P25. The effects of surface area and morphology of the BSO samples on different photocatalytic activity were also discussed.
1. Introduction
Ubiquitous organic pollutants are the most notorious contaminants in aquatic environments because of their huge volume of production from industries, slow biodegradation and decoloration, and toxicity.1–3Phenol and Rohdamine B (RhB) are often chosen as the model pollutants for they are the parent substrates of many organic pollutants, and in themselves the most common pollutants present in industrial wastewater. They are harmful if swallowed by human beings and animals, and cause irritation to the skin, eyes and respiratory tract. The carcinogenicity, reproductive and developmental toxicity, neurotoxicity and chronic toxicity towards humans and animals have been experimentally proved.4–6 During the last decades, photocatalytic technology for environmental protection procedures such as air purification, water disinfection, hazardous waste remediation and water decontamination have attracted extensive attention due to its mild reaction conditions and obviously depuration effects.7–11 Semiconductor photocatalysts with various morphologies, microstructures, and components have been developed.12–18 But photocatalysts with high photocatalytic activity (compared to the commercial TiO2, P25), good stability and low cost are still rare.Bismuth silicates is a conventional photorefractive, electro-optic material with good photoconductivity, which shows the high photocatalytic activity originates from the low recombination rate and high mobility of photogenerated electrons and holes.19–21 On the other hand, the (Bi2O2)2+ layers and (SiO4)4− tetrahedra in the crystal structure of BSO are slightly distorted. The distorted structure of (SiO4)4− tetrahedra may be in favor of the separation of the photogenerated holes and electrons.22–23 Besides these, Bi2O3 is also a good photocatalyst for water splitting and pollutant decomposing under visible light irradiation. So many research efforts have been focused on developing various morphologies of Bi2O3 or Bi2O3-containing complex and studying their optical properties.24–25Si stuff is also profuse in nature and inexpensive due to a content in the earth's crust of up to 25%. Despite BSO having these benefits mentioned above, only a few researchers have investigated the photocatalytic activity of bismuth silicate Bi2SiO5/Bi12SiO20 nanostructrues.22,23,28
Solid-state-reaction methods are the most common synthesis route for such materials.23,26,27 However, this route typically results in large agglomerated particles with low specific surface areas due to its high temperatures. Meanwhile, some different BSO phases such as Bi4Si3O12 and Bi12SiO20 were obtained spontaneously because of the metastable property of Bi2SiO5. Recently pure Bi2SiO5 nanosheets were synthesized by hydrothermal method,22 Zhang and co-workers have also reported hybrid Bi2SiO5 mesoporous microspheres with photoactivity in the degradation of RhB and phenol in water under simulated sunlight irradiation.28 Unfortunately, these methods not only relate to tedious synthetic procedures and high cost, but also revealed low photocatalytic performance. These drawbacks made it difficult for the large-scale industrial applications. It is well known that low-dimensional inorganic nanomaterials exhibit interesting and useful characteristics owing to quantum size effects and shape specialities. Some materials with beltlike and sheetlike nanostructures have attracted much attention,29–33 for example, Ga2O3 nanobelts,29Bi2GeO5 nanobelts,30TiO2 nanosheets,31MgO nanosheets,32NiO nanosheets,33 and various shapes of CoWO4, MnWO4, La2(MoO4)3.34 These low-dimensional nanocatalysts revealed better catalytic performance because their distinctive geometric structures are propitious to mobility of photogenerated electrons and holes. There is no report about the synthesis of Bi4Si3O12 nanobelts/nanosheets. It would be valuable to fabricate Bi4Si3O12 nanobelts/nanosheets with high photocatalytic activity. Successful synthetic strategies for the preparation of Bi4Si3O12 nanocrystals, especially for those nanorods, nanobelts, nanoflowers, and so on, are still a challenge.
Herein, we present a facile, self-assembly approach for mass production of Bi4Si3O12 nanobelts/nanosheets. The formation mechanism and morphology-controlled process of the samples were also discussed according to experimental data. Furthermore, both aqueous phenol and Rhodamine B (RhB) were employed as the probe reaction to test the photocatalytic activity of the as-synthesized samples under simulated sunlight irradiation. As a novel photocatalyst, the samples exhibited remarkable activity in the photocatalytic degradation of organic contaminants (aqueous phenol and azo dye RhB), which is higher than that of TiO2 (P25, Degussa). To the best of our knowledge, there is no report on the morphology controlled preparation of the BSO nanobelts/nanosheets. And the applications of the BSO nanobelts/nanosheets in the photocatalytic decomposition of organic pollutants was also investigated for the first time.
2. Experimental procedure
2.1 Catalyst preparation
Bismuth nitrate (Bi(NO3)3·5H2O) was purchased from Tianjin Kermel Chemical Reagents Development Centre (Tianjin, China). Tetraethyl orthosilicate ((C2H5O)4Si) and cetyltrimethyl ammonium bromide (CTAB) were provided by Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Ammonia (NH3·H2O) was obtained from Wuhan Chemical Reagent Co. All chemicals were analytical grade reagents and used without further purification. Deionized and doubly distilled water was also used in this work.A typical synthetic procedure was as follows: Bi(NO3)3 ·5H2O (2.43 g) and (C2H5O)4Si (0.52 g) were added into 20 mL of distilled water under stirring for 0.5 h, then 0.5 mmol of cetyltrimethyl ammonium bromide (CTAB) was also added in the above mixture. Under vigorous stirring, the pH value of the resultant solution was adjusted to 9 through the dripping of 25% NH3·H2O. After stirring for 0.5 h, the mixture was transferred into a Teflon-lined autoclave with a capacity of 100 mL. Then the autoclave was sealed and heated to 200 °C, then kept for 24 h under autogenous pressure. When the autoclave was cooled to room temperature, the precipitate was separated by filtration, washed with distilled water and absolute ethanol, and dried at 60 °C for 8 h. The other samples were prepared by a similar procedure, except for the different CTAB contents, and the samples obtained under various amounts of CTAB (x = 0.0, 0.25, 0.5, 1.0 mmol) were named S0, S1, S2, and S3, respectively.
2.2 Characterization
The crystalline structure of the as-prepared catalysts were characterized by power X-ray diffraction (XRD) employing a scanning rate of 0.05°/s in a 2θ range from 10° to 80°, in a Bruker D8 Advance using monochromatized Cu Kα radiation. The morphology of the catalysts were analyzed by the transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM), which were taken on a Tecnai G20 (FEI Co., Holland) TEM using an accelerating voltage of 200 kV. The TEM was connected to an energy dispersive spectrum analysis (EDS) system. The UV-vis DRS were collected using a Shimadzu UV-2450 spectrophotometer from 200 to 800 nm using BaSO4 as a reflectance standard. Specific surface areas of these samples were measured at 77 K by the BET method (N2 adsorption) using a Micromeritics ASAP 2020 instrument.2.3 Photocatalytic activity measurement
Both aqueous phenol and Rhodamine B (RhB) photodegradation were employed to test the photocatalytic activity of the as-prepared samples under simulated sunlight irradiation. The photodegradation experiments were performed under a 350 W Xe lamp as the light source to simulate the solar light irradiation.The degradation of phenol was carried out as follows: 0.05 g of each catalyst was suspended in 50 mL aqueous phenol (20 mg/L). The dispersed solution was kept in the dark for 60 min under stirring to reach adsorption/desorption equilibrium. At given time intervals, a 3 mL suspension was collected and centrifuged to remove the photocatalyst particles. Then, the adsorption spectrum of the centrifuged solution was recorded using a UV-vis spectrophotometer (UV-2450). The efficiency of degradation was calculated from a formula as follows:
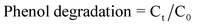
Besides, in order to determine the catalyst durability, the photocatalyst BSO was separated from aqueous solution after each run of reactions. The filtered catalyst was reused without any treatment in the subsequent recycling experiment.
3. Results and discussion
3.1 Characterization of catalysts
Fig. 1 shows the XRD patterns of the as-prepared BSO samples. It can be found that the XRD patterns of all samples present similar profiles. Six distinctive peaks at 21.12°, 27.38°, 32.55°, 43.03°, 44.89° and 54.97° were observed. These peaks match well with the (211), (310), (321), (422), (431) and (532) crystal planes of cubic Bi4Si3O12 (JCPDS no. 35-1007). The influence of surfactant CTAB on the crystalline phase was demonstrated through the changes of XRD patterns. As can be seen from Fig. 1, with the increasing of the concentration of surfactant CTAB, the Bi4Si3O12 increased gradually and the samples tend to be well-crystallized Bi4Si3O12 comparatively, while the other phase Bi2SiO5 (JCPDS no. 75-1483) decreased gradually.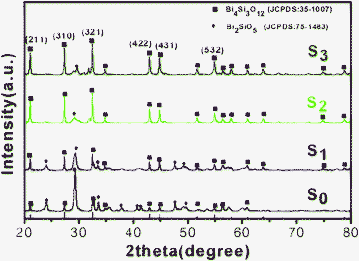 | ||
| Fig. 1 XRD patterns of the BSO samples: S0; S1; S2; S3. | ||
The morphologies of the synthesized samples were studied by TEM, which are shown in Fig. 2a–d. In order to investigate the effects of surfactants on the structure and morphology of the products, contrastive experiments were conducted. When the experiment was carried out in the absence of CTAB, the resulting products consist of a high yield of nanosheets, as shown in Fig. 2a. From Fig. 2b–c, nanosheets and nanobelts morphology are formed synchronously through the addition of the CTAB, and it is worth noted that the nanobelts morphology are synthesized with increasing amounts of CTAB. Moreover, the products consist almost entirely of the belt-like structure when 1.0 mmol of the surfactant CTAB is used, as shown in Fig. 2d. The typical widths of the nanobelts are in the range of about 50–100 nm, and their lengths extend to several micrometers. All of these results suggest that the relative amounts of CTAB can optimize the morphology of nanobelts.
 | ||
| Fig. 2 TEM images of the different BSO samples: (a) S0; (b) S1; (c) S2; (d) S3. | ||
The belt-like nanostructures achieved in the presence of CTAB (like sample S3) was further characterized with transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). Numerous nanobelts can be clearly observed in Fig. 3a. The nanobelts have widths of around 50–80 nm and lengths of several micrometers. The HRTEM image of an individual nanobelt is shown in Fig. 3c. The HRTEM image shows clear crystal lattices with d-spacing of ∼0.38 nm and ∼0.42 nm corresponding to (220) and (211) planes of cubic phase Bi4Si3O12, respectively. The nanobelt grows along the [220] direction. Meanwhile, FFT pattern (inset of Fig. 3c) exhibits a regular and clear diffraction spot array, which indicates that the nanobelt is single-crystalline nature. As shown in Fig. 3d, BSO belts is viewed along the [220] direction. The O atoms are red, the Si atoms are yellow and the Bi atoms are purple. Energy-dispersive X-ray (EDX) analysis (Fig. 3b) also shows that the atomic ratio of the Bi and Si is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in the S3 sample, which matches well with the element ratio of Bi4Si3O12. The elements of Cu and C are generated from the supporting carbon-coated copper meshes.
:3 in the S3 sample, which matches well with the element ratio of Bi4Si3O12. The elements of Cu and C are generated from the supporting carbon-coated copper meshes.
 | ||
| Fig. 3 The structure characterizations of S3: (a)TEM; (b)EDX spectrum; (c) HRTEM image; FFT pattern (inset of c); (d) The crystal structure model of Bi4Si3O12. The O atoms are red, the Si atoms are yellow and the Bi atoms are purple. | ||
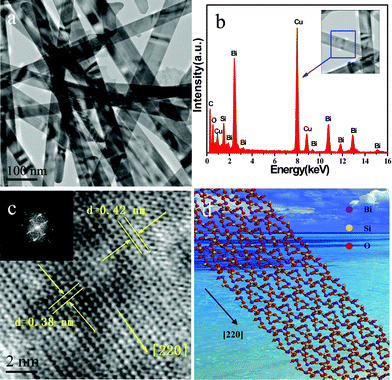
According to the TEM images of the samples, a schematic diagram of the proposed growth mechanism is shown in Fig. 4. In the CTAB-free hydrothermal process, there are no organic molecules around the circumference of the BSO nuclei, these BSO nanoparticles will attach to each other. Consequently, BSO nanosheets S0 are obtained (in Fig. 4). When a small amount of CTAB was added, capsules of CTAB are generated in the solution. These capsules can cause BSO nuclei to change the shape and size of the primary clusters, because the hydrophobic interactions and Van Der Waals attraction between surfactant molecules and adjacent nuclei lead to assembly among these BSO nanoparticles. Thus, longitudinal growth is dominant, and subsequently the constructional BSO nanoparticles grows through aggregation in a certain direction to form nanobelts, except for the partial nanosheets (S1 and S2 in Fig. 4). When CTAB was introduced in appropriate amounts, the initially formed nanoparticles were relatively unstinted in dimension, and well dispersed in the solution. The selective adsorption of CTAB on specific crystal faces promotes BSO crystals to grow along the [220] direction forming the perfect nanobelts (sample S3 in Fig. 4). Possible functions for CTAB in the present synthetic method mainly act as soft-templates for the formation of 1D BSO nanobelts as well as stabilize the 1D nanobelts. The present work is mainly focused on the control of BSO morphology by adjusting the concentration of surfactant CTAB. The detailed formation mechanism of BSO nanobelts/nanosheets in this hydrothermal process needs further investigation.
 | ||
| Fig. 4 Schematic illustration of the proposed growth mechanism of BSO nanostructures. | ||
3.2 Photocatalytic activity
The photocatalytic activities of the as-prepared BSO samples were evaluated firstly by the degradation of phenol. Fig. 5 shows the concentration changes of phenol at 269 nm as a function of irradiation time during the degradation process in aqueous solution in the presence of BSO samples. For comparison, the photocatalytic degradation rate of phenol in the absence of the photocatalyst or the light were also conducted respectively, and the results showed that only a small quantity of phenol was degraded (inset of Fig. 5). For Degussa P25, only 23.1% of phenol was decomposed. Obviously, all as-prepared BSO samples increased the percentage of phenol (20 mg/L) degradation (for S0, 62.7% degradation, whereas after the modification of CTAB, 98.5% degradation was observed for S1, 57.3% for S2, and 37.1% for S3 under identical conditions) after Xe light irradiation for 180 min, which demonstrates the as-prepared BSO photocatalysts exhibited higher photocatalytic performance for organic pollutants degradation (compared to the commercial TiO2, P25). It is interesting to note that 98.5% of the phenol in the solution is decomposed over S1. Fig. S1† shows the temporal evolution of the spectra during the photodegradation of phenol (initial concentration: 20 mg/L, 50 mL) in the presence of the S1 sample under simulated sunlight illumination. It is clearly seen that the absorption peaks at 269 nm corresponding to phenol diminished gradually.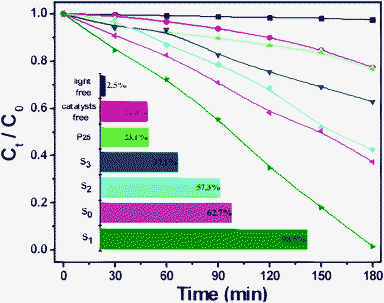 | ||
| Fig. 5 Concentration changes of phenol (20 mg/L) at 269 nm as a function of irradiation time in aqueous solution in the presense of BSO samples (50 mg). | ||
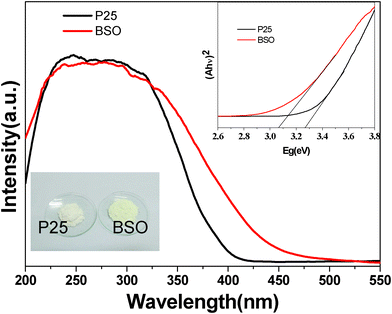
In order to further investigate the photocatalytic activity for organic dyes of the as-prepared samples, S1 was chosen as a representative sample. Fig. 6 shows the decoloration rate of RhB over S1 and P25 TiO2 powders at the same time interval. The changing of the concentration proves that photocatalytic activity of S1 is higher than that of P25 TiO2 under simulated sunlight irradiation. To clarify the reason for the higher photocatalytic activity of S1 compared to P25, we further measured the UV-vis diffuse reflectance absorption spectra UV-vis DRS of S1 and P25 using a Shimadzu UV-2450 spectrophotometer. Fig. 7 illustrates the UV-vis DRS of S1 and P25 TiO2. It can be seen that S1 can absorb visible light in the wavelength range of about 400–500 nm, which is in agreement with the pale yellow color of the synthesized samples. Compared with P25 TiO2 (∼3.26 eV), the band gap energies of S1 was calculated to be ∼3.07 eV. Therefore, the better activity of S1 may be ascribe to an extensive absorbance in visible light.
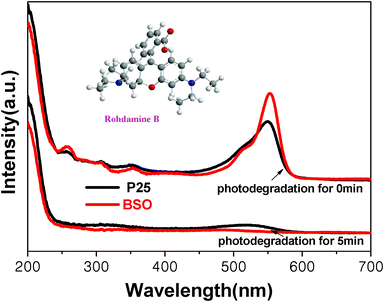 | ||
| Fig. 6 UV-vis curves of degradation of RhB over the BSO and P25 TiO2 at the same time interval. | ||
In the present work, the addition of different amounts CTAB results in a controllable morphology of the bismuth silicate nanobelts/nanosheets, and the BSO samples shows good photocatalytic performance in organic pollutants decomposition. BSO nanobelts/nanosheets structures exhibit a lower electron-hole recombination rate due to the following reasons: (i) greater charge mobility in the nanobelts/nanosheets, which is enabled along the longitudinal dimension of the crystals; (ii) low-dimension nanobelts/nanosheets could enhance charge separation due to trapping of photogenerated electrons by chemisorbed molecular O2.35 Moreover, the differences in activity of the various BSO may be related to their distinct morphologies. The Brunauer–Emmett–Teller adsorption analysis showed that the BET surface areas of the synthesized S0, S1, S2 and S3 samples were 9.43 m2/g, 13.85 m2/g, 5.7 m2/g and 6.23 m2/g, respectively. The surface area of the sample S1 is larger than the other samples. The significant difference in the surface area should be relate to their activities. It is well-known that a higher surface area increases the number of active sites, resulting in a higher photocatalytic activity.36–37 Therefore, sample S1 displays the most promising photoactivity than other BSO samples. Some unknown positive effects maybe exist between nanobelts and nanosheets.
As a potential photocatalyst for degradation of organic contaminants, the stability of the catalyst is one of key factors for practical use. Here, in order to test the stability of the BSO sample, the cyclic photodegradation of RhB under simulated sunlight irradiation in the presence of the sample S1 shown in Fig. 8. The result reveals that the BSO nanobelts exhibits excellent photostability and shows no noticeable loss of photocatalytic activity after 6 cycles. The results demonstrate that the nanostructured BSO is a promising green photocatalyst.
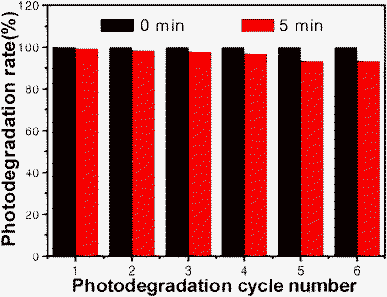 | ||
| Fig. 8 The cyclic photodegradation of RhB(10−5 M) under simulated sunlight irradiation in the presence of the sample BSO(S1: 50 mg). | ||
4. Conclusions
In summary, Bi4Si3O12 nanobelts/nanosheets were successfully synthesized by a surfactant-templated hydrothermal process for the first time. Photocatalytic evaluation via the decomposition of phenol and RhB under simulated sunlight irradiation, revealed that these BSO samples exhibited excellent photocatalytic performances compared to P25. S1 with the morphology of nanobelts and nanosheets possesses the best photocatalytic activity because of a larger surface area and special morphology. And the UV-vis DRS also attests that the as-prepared nanomaterials have strong absorption in visible light. Furthermore, the novel BSO materials showed good stability.Acknowledgements
The Project was sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry. This work was also supported by National Natural Science Foundation of China (20803096, 21073238), NSF of Hubei Province (Distinguished Young Investigator Grant 2010CDA082) and National Basic Research Program of China (Grant No: 2011CB211704).References
- H. Kyung, J. Lee and W. Choi, Environ. Sci. Technol., 2005, 39, 2376 CrossRef CAS.
- W. Y. Dong, C. W. Lee, X. C. Lu, Y. J. Sun, W. M. Hua, G. S. Zhuang, S. C. Zhang, J. M. Chen, H. Q. Hou and D. Y. Zhao, Appl. Catal., B, 2010, 95, 197 CrossRef CAS.
- V. I. Panvulescu and H. Garcia, Catalysis, 2011, 23, 204 Search PubMed.
- K. Kadirvelu, C. Karthika, N. Vennilamani and S. Pattabhi, Chemosphere, 2005, 60, 10097 CrossRef.
- Z. Chen, D. Li, W. Zhang, Y. Shao, T. Chen, M. Sun and X. Fu, J. Phys. Chem. C, 2009, 113, 4433 CAS.
- R. Jain, M. Mathur, S. Sikarwar and A. Mittal, J. Environ. Manage., 2007, 85, 956 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- G. Liu, Z. G. Chen, C. L. Dong, Y. N. Zhao, F. Li, G. Q. Lu and H. M. Cheng, J. Phys. Chem. B, 2006, 110, 20823 CrossRef CAS.
- A. H. Lu, Y. Li, M. Lv, C. Q. Wang, L. Yang, J. Liu, Y. H. Wang, K. H. Wong and P. K. Wong, Sol. Energy Mater. Sol. Cells, 2007, 91, 1849 CrossRef CAS.
- J. G. Yu, Y. R. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984 CrossRef CAS.
- F. A. Momani and N. Jarrah, Environmental Technology, 2009, 30, 1085 CrossRef.
- J. G. Yu, Y. R. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984 CrossRef CAS.
- X. F. Chen, X. C. Wang and X. Z. Fu, Energy Environ. Sci., 2009, 2, 872 CAS.
- Y. B. Chen, L. Z. Wang, G. Q. Lu, X. D. Yao and L. J. Guo, J. Mater. Chem., 2011, 21, 5134 RSC.
- T. F. Zhou and J. C. Hu, Environ. Sci. Technol., 2010, 44, 8898 Search PubMed.
- L. Kong, Z. Jiang, T. C. Xiao, L. F. Lu, M. O. Jones and P. P. Edwards, Chem.Comm., 2011, 47, 5512 RSC.
- S. Y. Yang, P. N. Zhu, A. S. Nair and S. Ramakrishna, J. Mater. Chem., 2011, 21, 6541 RSC.
- X. X. Yu, S. W. Liu and J. G.Yu, Appl. Catal., B, 2011, 104, 12 CrossRef CAS.
- A. E. Attard, J. Appl. Phys., 1991, 69, 44 CrossRef CAS.
- S. C. Abrahams, J. L. Bernstein and C. Svensson, J. Chem. Phys., 1979, 71, 788 CrossRef CAS.
- Y. T. Fei, S. J. Fan, R. Y. Sun and J. Y. Xu, J. Mater. Sci. Lett., 2000, 19, 893 CrossRef CAS.
- R. G. Chen, J. H. Bi, L. Wu, W. J. Wang, Z. H. Li and X. Z. Fu, Inorg. Chem., 2009, 48, 9072 CrossRef CAS.
- C. H. He and M. Y. Gu, Scr. Mater., 2006, 55, 481 CrossRef CAS.
- L. Zhou, W. Z. Wang, H. L. Xu, S. M. Sun and M. Shang, Chem.–Eur. J., 2009, 15, 1776 CrossRef CAS.
- Y. Wang, S. K. Li, X. R. Xing, F. Z. Huang, Y. H. Shen, A. J. Xie, X. F. Wang and J. Zhang, Chem. Eur. J., 2011, 17, 4802 CrossRef CAS.
- Z. G. Zhang, X. F. Wang, Q. Q. Tian and C. L. Yu, Science of Sintering, 2009, 41, 83 CrossRef CAS.
- M. Ishii, K. Harada, Y. Hirose, N. Senguttuvan, M. Kobayashi, I. Yamaga, H. Ueno, K. Miwa, F. Shiji, F. Yiting, M. Nikl and X. Q. Feng, Opt. Mater., 2002, 19, 201 CrossRef CAS.
- L. Zhang, W. Z. Wang, S. M. Sun, J. H. Xu, M. Shang and J. Ren, Appl. Catal., B, 2010, 100, 97 CrossRef CAS.
- L. C. Tien, W. T. Chen and C. H. Ho, J. Am. Ceram. Soc., 2011, 94, 3117 CrossRef CAS.
- R. G. Chen, J. H. Bi, L. Wu, Z. H. Li and X. Z. Fu, Cryst. Growth Des., 2009, 9, 1775 CAS.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 11, 4078 CrossRef.
- J. C. Hu, K. K. Zhu, L. F. Chen, C. Kubel and R. Richards, J. Phys. Chem. C, 2007, 111, 12038 CAS.
- J. C. Hu, K. K. Zhu, L. F. Chen, H. J. Yang, Z. Li, A. Suchopar and R. Richards, Adv. Mater., 2008, 20, 267 CrossRef CAS.
- T. D. Nguyen, C. T. Dinh and T. O. Do, Nanoscale Res. Lett., 2011, 3, 1861 CAS.
- (a) N. Q. Wu, J. Wang, D. N. Tafen, H. Wang, J. G. Zheng, J. P. Lewis, X. G. Liu, S. S. Leonard and A. k. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679 CrossRef CAS; (b) B. Ohtani, Y. Ogawa and S. Nishimoto, J. Phys. Chem. B, 1997, 101, 3746 CrossRef CAS.
- W. W. Wang, Y. J. Zhu and L. X. Yang, Adv. Funct. Mater., 2007, 17, 59 CrossRef CAS.
- A. Fumiaki, Y. Akira, N. Kohei, O. Masatoshi and O. Bunsho, J. Am. Chem. Soc., 2008, 130, 17650 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00188d |
| This journal is © The Royal Society of Chemistry 2011 |