Two mononuclear iron(II) complexes of 4-phenylpyrazole-5-carbaldehyde derived ligands are stabilised in different spin states†
Juan
Olguín
,
Guy N. L.
Jameson
and
Sally
Brooker
*
Department of Chemistry and The MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin 9054, New Zealand. Fax: +64 3 479 7906; Tel: +64 3 479 7919E-mail: sbrooker@chemistry.otago.ac.nz
First published on 18th July 2011
Abstract
The polydentate acyclic ligand 4-phenylpyrazole-5-carbaldehyde azine, H2L1, has been prepared and characterised. A second 4-phenylpyrazole-5-carbaldehyde derived ligand, H2L2, was prepared in situ by condensation with o-phenylendiamine. Two mononuclear iron(II) complexes, [FeII(HL2)(MeOH)(NCSe)]·H2O and [FeII(H2L1)2](BF4)2·solvents, have been synthesised from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:3 M:L reactions, respectively. No dinuclear, helical, species were obtained from 2:3 M:L reactions regardless of the solvent employed (MeOH, MeCN or NO2Me). An X-ray structure determination on the latter complex reveals that it is low spin at 90 K. Mössbauer spectra confirm this, and show that the former complex is, in contrast, high spin, even at 4.6 K.
:1 and 2:3 M:L reactions, respectively. No dinuclear, helical, species were obtained from 2:3 M:L reactions regardless of the solvent employed (MeOH, MeCN or NO2Me). An X-ray structure determination on the latter complex reveals that it is low spin at 90 K. Mössbauer spectra confirm this, and show that the former complex is, in contrast, high spin, even at 4.6 K.
Introduction
Spin crossover (SCO) between low-spin (LS) and high-spin (HS) states in octahedral d4–d7 first row transition metal ions is an important phenomenon due to the possible future application of these materials in molecular switches, data storage and other molecular devices.1,2 The interchange between the magnetic states can be promoted by an external stimulus such as change in temperature or pressure, light irradiation or application of a magnetic field.The most widely studied SCO-active transition metal is iron(II).3 SCO-active iron(II) complexes that show the highly desirable property of thermal hysteresis4 usually have high cooperativity between the iron(II) centres. In mononuclear complexes this is normally a result of intermolecular hydrogen bond interactions, anion-π and/or π–π interactions,5 whereas in polynuclear species it can also be mediated by bridging ligands.1
Both mononuclear [MII(PAA)2] and dinuclear helical complexes [MII2(PAA)3] (MII = Fe and Ni) were prepared by Stratton and Busch,6 illustrating the versatility of the pyridine-2-carbaldehyde azine ligand, PAA (Fig. 1A). Both types of iron(II) complexes were LS. In order to reduce the ligand field experienced by the iron(II) centres and increase the chances of observing SCO, Kojima and co-workers7 designed a series of substituted imidazole-4-carbaldehyde azine ligands (Fig. 1B). In addition to imidazole offering a weaker ligand field than pyridine, it offers another advantage, the possible engagement of the ‘spare’ NH groups in hydrogen bond interactions, increasing the cooperativity within the crystal lattice and the chances of observing hysteresis loops. These authors reported the synthesis of families of mononuclear, and dinuclear helical, iron(II) complexes. Mononuclear complexes were obtained when the protic solvent methanol was used whereas dinuclear complexes were obtained when nitromethane or acetonitrile was used; the Fe:L ratio did not control the outcome. Four of the dinuclear helical complexes underwent a ‘half’ SCO from [HS-HS] to [HS-LS] and two exhibited small hysteresis loops (4–7 K).
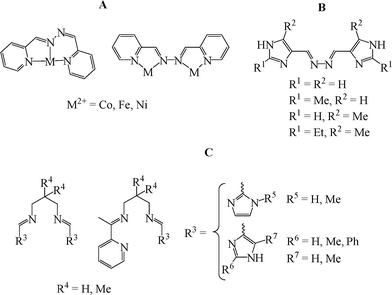 | ||
| Fig. 1 Schematic representation of: (A) the flexidentate ligand PAA and the two coordination modes it has been shown, by Stratton and Busch,6 to adopt; (B) the imidazole-4-carbaldehyde azine ligands synthesised by Kojima and co-workers;7 (C) the tetradentate symmetric and asymmetric Schiff base ligands synthesised by Tuchagues, Bréfuel and co-workers.8 | ||
Tuchagues, Bréfuel and co-workers8 used imidazole 4- and 2-carbaldehydes to synthesise, in situ, a range of symmetric and asymmetric tetradentateSchiff base ligands (Fig. 1C). The ligand solutions were reacted 1:1 with [FeII(ECN)2(MeOH)4] (E = S or Se), resulting in a series of monometallic complexes of the type [FeII(L)(ECN)2]. Only the asymmetric pyridine-imidazole complexes underwent SCO, at around room temperature. A few of these complexes exhibited small hysteresis loops.
Given our ongoing interests in pyrazole-based ligands,9,10 we decided to study the effect of changing the position of the ‘spare’ NH group in the above ligands by replacing imidazole with pyrazole. Pyrazole is also known to impose a suitable ligand field for SCO in iron(II).11 Like imidazole, the pyrazole ring possesses two types of nitrogen donors, an imine-like C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and an amine like NH donor, however, in pyrazole these nitrogen atoms are located in adjoining, instead of alternating, positions. This should impact on the nature of the packing interactions and on the magnetic behaviour of the resulting iron(II) complexes.
N and an amine like NH donor, however, in pyrazole these nitrogen atoms are located in adjoining, instead of alternating, positions. This should impact on the nature of the packing interactions and on the magnetic behaviour of the resulting iron(II) complexes.
In this study, the pyrazole-based head unit 4-phenylpyrazole-5-carbaldehyde10 was employed to generate two types of ligand, the ‘flexidentate’ ligand H2L1 and the tetradentate ligand H2L2 (Fig. 2), which are analogues of the above azine and tetradentate ligands respectively (Fig. 1B and 1C). The iron(II) coordination chemistry of these ligands is reported herein.
2·solvents, and of the mononuclear complex of HL2, [FeII(HL2)(MeOH)(NCSe)]·H2O; i. N2H4·H2O, MeOH reflux; ii. ½ o-phenylendiamine, [FeII(Py)4(SeCN)2], MeOH; iii. ½ FeII(BF4)2·6H2O, MeNO2.](/image/article/2011/RA/c1ra00190f/c1ra00190f-f2.gif) | ||
| Fig. 2 Synthesis of ligand H2L1 and mononuclear complex [FeII(H2L1)2](BF4)2·solvents, and of the mononuclear complex of HL2, [FeII(HL2)(MeOH)(NCSe)]·H2O; i. N2H4·H2O, MeOH reflux; ii. ½ o-phenylendiamine, [FeII(Py)4(SeCN)2], MeOH; iii. ½ FeII(BF4)2·6H2O, MeNO2. | ||
Results and discussion
The ligand H2L1 was synthesised by refluxing a suspension of 4-phenylpyrazole-5-carbaldehyde10 with hydrazine hydrate in methanol, then concentrating the yellow solution and cooling it to precipitate the desired ligand as a bright yellow solid in 80% yield. After drying under vacuum the ligand was characterised by 1H NMR spectroscopy and used without further purification. The IR spectrum of H2L1 showed the expected CN imine stretch at 1630 cm−1.
Kojima and co-workers7 showed that the synthesis of mono- and di-nuclear complexes of the analogous imidazole-4-carbaldehyde-based azine ligands (Fig. 1B) is solvent dependent. The dinuclear helical Fe2L3 complexes were synthesised from aprotic polar solvents, such as nitromethane and acetonitrile, at 0 °C, whereas when methanol was used at room temperature the mononuclear FeL2 complexes were obtained.
We first attempted the synthesis of the dinuclear complexes by adding two equivalents of solid FeII(BF4)2·6H2O to a suspension of 3 equivalents of the ligand H2L1 in nitromethane under a nitrogen atmosphere at room temperature. The resulting deep purple solution was stirred for 2 h, filtered and then vapour diffused with diethyl ether in air. After a few days purple crystals, unsuitable for X-ray crystallography, were obtained. After drying in vacuo, microanalysis of the purple microcrystalline solid was in agreement with the formation of the mononuclear 1:2 Fe:L complex [FeII(H2L1)2](BF4)2·2.5H2O, despite the excess of iron used (2:3 Fe:L). The IR spectrum has two CN stretches, at 1620 and 1606 cm−1, consistent with the asymmetric coordination mode found in the mononuclear complex, but not the dinuclear helical complex. The deep purple colour of the sample indicated that the iron(II) centre is LS. The 1H NMR spectrum is consistent with this, with sharp, well defined resonances typical of diamagnetic samples (Fig. 3). Moreover the number of resonances observed is correct for the pseudo C2 symmetric mononuclear complex. The LS state of the complex was confirmed by Mössbauer spectroscopy (Table 1, Fig. S2, ESI†) at room temperature; hence this complex does not undergo thermal SCO below 300 K.
2·2.5H2O in CD3CN, 298 K.](/image/article/2011/RA/c1ra00190f/c1ra00190f-f3.gif) | ||
| Fig. 3 1H NMR spectrum of [FeII(H2L1)2](BF4)2·2.5H2O in CD3CN, 298 K. | ||
In another attempt to obtain the dinuclear helical complex, an excess of iron(II) salt (2.5:3 Fe:L, 25% excess for desired product) was used under otherwise identical reaction conditions. In this case, after a few days of diethyl ether vapour diffusion, the purple crystals obtained were suitable for X-ray crystallography (see later), confirming the formation of the mononuclear complex [FeII(H2L1)2](BF4)2·2.5H2O, despite the excess iron(II) present (67% excess for this product).
The first reaction was therefore repeated, but at 0 °C. The resulting purple nitromethane solution was stirred for two hours before a large volume of diethyl ether was added by cannula, causing a purple solid to precipitate. This was filtered off under nitrogen, dried in vacuo and shown to be the mononuclear complex by microanalysis. The use of methanol as the solvent at 0 °C resulted in the same product.
The low solubility of the ligand in acetonitrile required the addition of few millilitres of methanol or nitromethane to the reaction mixture: in both cases the mononuclear complex was obtained. The use of anhydrous iron(II) acetate as the starting material in the 2:3 Fe:L reactions resulted in very soluble products that could not be cleanly isolated.
To summarise, in our hands careful control of stoichiometry, solvent and temperature did not lead to a dinuclear helical complex of H2L1. Rather, the mononuclear complex was consistently obtained, a result which contrasts with those obtained with the imidazole and pyridine ligand analogues.
The tetradentate ligand H2L2 was formed in situ, by a 2:1 reaction of 4-phenylpyrazole-5-carbaldehyde10 and o-phenylendiamine in a 1:1 methanol/acetonitrile mixture at reflux for 30 min, prior to complexation, as attempts to isolate it cleanly were not successful (1H NMR spectra revealed a mixture of H2L2, mono imine and unreacted aldehyde, Fig. 4).
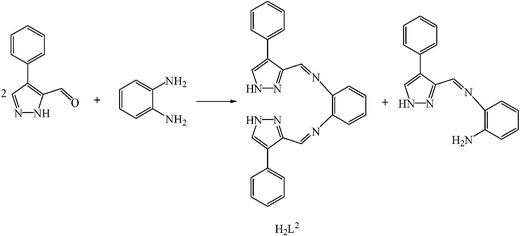 | ||
| Fig. 4 Condensation products identified by 1H NMR spectroscopy of the 2:1 reaction of 4-phenylpyrazole-5-carbaldehyde and o-phenylendiamine. | ||
The in situ ligand reaction solution was allowed to cool down to room temperature before it was degassed and one equivalent of solid [FeII(Py)4(ECN)2] (ES or Se)12 added under nitrogen. For SCN a dark-red solution resulted whereas for SeCN a yellow solution was obtained. Both solutions were concentrated to half the volume and subjected to diethyl ether vapour diffusion, in an H-tube under argon. After a few weeks a red (SCN) and a white (SeCN) solid were filtered off under argon and dried in vacuo. The red SCN product was intractable, possibly due to higher air sensitivity; however, the white SeCN product gave microanalysis results which were in agreement with the mononuclear complex [FeII(HL2)(MeOH)(NCSe)]·H2O. This suggests that spontaneous mono deprotonation of the ligand has occurred, in the absence of added base (Fig. 2). This is not surprising as it is known that upon coordination the pKa of the pyrazole ring decreases,13 and added to this there are structural reasons that probably favour this, with the remaining proton likely forming a H-bond between the two facing pyrazole rings in the equatorial plane. It is curious that in this instance only one of the two SeCN anions initially coordinated to the iron(II) in the reagent remains in the product; this may be due to the mono deprotonation of the ligand leading to only one SeCN being required to balance the charge. The result is an N5O coordination environment, with the oxygen donor coming from methanol.
The IR spectrum showed the NC stretch at 1604 cm−1, whilst the band at 2064 cm−1 is indicative of the N-bonded SeCN moiety. The light colour of the solid strongly indicated that this is a HS iron(II) complex. Mössbauer spectroscopy data collected at 4.6 K confirmed the HS state and that no thermal SCO occurs for this complex (Table 1 and Fig. S1†). This is not particularly surprising as usually an N6 environment is required for SCO in iron(II).14
X-Ray crystallography
The complex [FeII(H2L1)2](BF4)2·¼(Et2O)·¼(H2O)·MeOH crystallises in the C2/c space group (Fig. 5) with the asymmetric unit comprising the entire compound. One tetrafluoroborate anion and two phenyl rings at the back of the pyrazole ring are disordered (see cif file for more details†). The iron(II) centre is coordinated to two H2L1 ligands. Each ligand is coordinated in an asymmetric tridentate mode (only one of the two nitrogen donors of the azine group is coordinated), resulting in adjacent 5- and 6-membered chelate rings. The average Fe–N bond length of 1.945 Å is in agreement with the assigned LS-state, and with the parameters found for the only LS mononuclear complex of the imidazole series.152·¼(Et2O)·¼(H2O)·MeOH. The hydrogen atoms (except for NH) and solvent molecules have been omitted for the sake of clarity. The smaller occupancy carbon atoms of the disordered phenyl groups are not shown.](/image/article/2011/RA/c1ra00190f/c1ra00190f-f5.gif) | ||
| Fig. 5 View of the cation of [FeII(H2L1)2](BF4)2·¼(Et2O)·¼(H2O)·MeOH. The hydrogen atoms (except for NH) and solvent molecules have been omitted for the sake of clarity. The smaller occupancy carbon atoms of the disordered phenyl groups are not shown. | ||
Both of the pyrazole rings in each ligand strand remain protonated and hydrogen bond to the BF4 anions. Therefore each tetrafluoroborate anion is ‘sandwiched’ by two complex cations (Fig. S1 and Table S2, ESI†). These interactions may be contributing to the exclusive formation of the mononuclear complex, as this coordination mode probably maximizes the number of hydrogen bond interactions (Fig. 6).
2·¼(Et2O)·¼(H2O)·MeOH showing the BF4 anions (spacefill) accommodated between complex cations, maximising the H-bond interactions.](/image/article/2011/RA/c1ra00190f/c1ra00190f-f6.gif) | ||
| Fig. 6 Unit cell of complex [FeII(H2L1)2](BF4)2·¼(Et2O)·¼(H2O)·MeOH showing the BF4 anions (spacefill) accommodated between complex cations, maximising the H-bond interactions. | ||
Conclusions
We have successfully synthesised and characterised the pyrazole-based azine ligand H2L1, an analogue of the imidazole-based azine system employed by Kojima and coworkers (Fig. 1B). Coordination of H2L1 to Fe(BF4)2·6H2O consistently resulted in a mononuclear complex stabilized in the LS state, regardless of the solvent, temperature and metal to ligand ratio employed.To date, unlike in the imidazole system, the dinuclear helical complex of H2L1 has been elusive. That the mononuclear complex is low spin and significantly stabilized in the solid state by extensive hydrogen bond interactions likely facilitates this. Certainly it is clear that changing the position of the ‘spare’ NH group in the five membered ring heterocycle, from alternating in imidazole to adjacent in pyrazole, has had a big impact on the reaction outcomes.
The tetradentate pyrazole-based ligand H2L2 was synthesised in situ and reacted with [FeII(Py)4(NCSe)2] under an inert atmosphere, resulting in the mononuclear N5O coordinated complex [FeII(HL2)(MeOH)(NCSe)]·H2O, stabilised in the HS state. Attempts to synthesise the SCN analogue resulted in highly air-sensitive products which have therefore not been pursued further. It had been hoped that such [FeII(HL2)XY] complexes might, on further deprotonation, be useful, magnetically interesting, monometallic building blocks for subsequently generating families of heterometallic complexes akin to [FeII(NiIIL2)3]2+, however, in light of these results it appears that we will be better served by a ‘one pot’ approach,10 a route we are actively pursuing.
Experimental
General
[Fe(Py)4(SeCN)2] was synthesised following the procedure described as for the SCN analogue.12Solvents and reagents (reagent grade) were purchased from commercial suppliers and used without further purification except for the HPLC grade solvents used in the synthesis of the complex [FeII(HL2)(MeOH)(NCSe)]·H2O.Elemental analyses were carried out by the Campbell Microanalytical Laboratory at the University of Otago. Infrared spectra were recorded over the range 4000–400 cm−1 in a Bruker Alpha FT-ATR with an Alpha-P module. ESI mass spectra were recorded on a Bruker MicrOTOFQspectrometer by Mr Ian Stewart. 1H NMR spectra were recorded on a Varian INOVA-400 NMR spectrometer at 25 °C.
57Fe Mössbauer spectra were recorded by Dr Guy N. L. Jameson at the University of Otago. Approximately 40 mg of sample was placed in a nylon sample holder (12.8 mm diameter, 1.6 mm thickness) with Kapton windows. Mössbauer spectra were measured on a Mössbauer spectrometer from SEE Co. (Science Engineering & Education Co., MN) equipped with a closed cycle refrigerator system from Janis Research Co. and SHI (Sumitomo Heavy Industries Ltd.). Data were collected in constant acceleration mode in transmission geometry with an applied field of 47 mT perpendicular to the γ-rays. The zero velocity of the Mössbauer spectra refers to the centroid of the room temperature spectrum of a 25 μm metallic iron foil. Analysis of the spectra was conducted using the WMOSS program (SEE Co., formerly WEB Research Co. Edina, MN).
Crystal data for [FeII(H2L1)2](BF4)2·¼(Et2O)·¼(H2O)·MeOH: C42H39B2F8FeN12O1.50, M = 965.32 g mol−1, monoclinic, a = 23.047(10) Å, b = 23.329(10) Å, c = 17.388(7) Å, β = 95.974(13)°, T = 84(2)K, space groupC2/c, volume 9298(7) Å3, Z = 8, 25 950 reflections measured, 7138 unique (Rint = 0.1815) which were used in all calculations; R1 = 0.1208 (4σ) and wR2 = 0.3960 (all data; the crystal was a very weak diffractor hence the high value). The X-ray data (see also Table S1, ESI†) were collected on a Bruker APEX II area detector diffractometer at the University of Otago using graphite-monochromated MoKa radiation (λ = 0.71073 Å). The data were corrected for Lorentz and polarization effects, and semi-empirical absorption corrections (SCALE) were applied. The structures were solved by direct methods (SHELXS-97) and refined against all F2 data (SHELXL-97).16Hydrogen atoms were inserted at calculated positions and rode on the atoms to which they were attached, except for those attached to methanol and water molecules that were found in the map. All non-hydrogen atoms were made anisotropic except for: the half occupancy diordered methanol solvent molecules (O60-C60 and O70-C70), the quarter occupancy diethyl ether molecule (O50-C51-C52; O50 sits on a two-fold axis) and H2O (O80) molecule, the disordered BF4 (B2 F21-F24vs. B25 F24, F26-F28), and the non-pivot atoms of the three of the four parts of two disordered phenyl rings (0.75 occupancy C38-C42 and 0.25 occupancy C100-C104 and C90-C94). CCDC 796801 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
4-Phenylpyrazole-5-carbaldehyde azine (H2L1)
To a suspension of 4-phenylpyrazole-5-carbaldehyde10 (75.0 mg, 0.30 mmol) in 20 mL of HPLC methanol, a solution of hydrazine hydrate (5.2 mL of a 28 mM solution in HPLC methanol, 0.15 mmol) was added at once and refluxed for 2 h, before the solution was cooled down to room temperature and placed in the fridge. After a few hours a bright yellow solid was present, it was filtered and dried under vacuum and used without further purification (45.0 mg, 0.13 mmol, 90% yield). 1H NMR 400 MHz (CDCl3 @ 7.26 ppm): 1H NMR 400 MHz (CDCl3 @ 7.26 ppm): 0.95 (s, 4H, CH3OH), 3.49 (s, 12H, CH3OH) 7.39–7.49 (m, 10 H, Ph-Pyr), 7.79 (s, 2H, 3-Pyr), 8.68 (s, 2H, CHNimine), 10.88 (s br, 2H, NHpyrazole). IR (ATR) cm−1: 3119 (br), 2923 (m), 2813 (m), 1630 (m), 1451 (m), 1110 (m), 1021 (s), 943 (s), 829 (s), 761 (s), 699 (s). ESI(+) MSm/zMeOH: calcd for [M+H]+ 341.1509 found 341.1497.
[FeII(H2L1)2](BF4)2·2.5H2O:
To a deoxygenated suspension of H2L1 (0.12 g, 0.34 mmol) in 30 ml of nitromethane, solid Fe(BF4)2·6H2O (0.077 g, 0.23 mmol) was added under an argon atmosphere, the resulting dark purple solution was stirred for 2 h before filtered and placed in a diethyl ether jar in air. After a few days purple crystals were obtained, filtered and dried under vacuum (64.0 mg, 0.068 mmol, 40% yield). Microanalysis: Calcd for [FeII(C20H16N6)2](BF4)2·2.5H2O C 50.29, H 3.90, N 17.60, found C 50.23, H 3.74, N 17.53. 1H NMR 400 MHz (CD3CN @ 1.94 ppm): 7.42–7.67 (m, 10 H, Ph-Pyr), 7.90 and 7.96 (s, 1H each one, 3-Pyr), 8.49 (s, 1H, CHNimine uncoordinated), 10.28 (s, 1H, CHNimine coordinated), 11.58 (s br, 2H, NHpyrazole). IR (ATR) cm−1: 3213 (br), 3125 (w), 3019 (w), 1620 (m), 1606 (m), 1391 (m), 1364 (m), 1052 (vs), 1004 (vs),993 (vs), 948 (s), 728 (s), 694 (s). ESI(+)MS (m/z) MeOH/MeCN: Calcd for [Fe(C20H16N6)2]+ 736.2218, found 736.2066.
[FeII(HL2)(MeOH)(NCSe)]·H2O
4-Phenylpyrazole-5-carbaldehyde 10 (69.0 mg, 0.40 mmol) and o-phenylendiamine (21.10 mg, 0.20 mmol) were suspended in 30 mL of a 1:1 methanol/acetonitrile mixture and refluxed for 0.5 h, before the solution was cooled down at room temperature and the yellow solution deoxygenated. Solid [Fe(Py)4(SeCN)2] (0.11 g, 0.2 mmol) was added under an argon atmosphere. The resulting yellow solution was stirred for 2 h at room temperature before it was concentrated to half the volume and placed in an H-tube for diethyl ether vapour diffusion under an argon atmosphere. After a few weeks a white powder was present. It was filtered off under argon and dried under vacuum (45.0 mg, 0.07 mmol, 36% yield). Microanalysis: Calcd for [FeII(C26H22N6)(CH3OH)(NCSe)]·H2O C 52.36, H 3.92, N 15.26, found C 52.38, H 3.65, N 14.65. IR (ATR) cm−1: 3204 (br), 2928 (m), 2064 (m), 1604 (m), 1450 (m), 1341 (m), 1304 (m), 1092 (s), 740 (vs), 695 (s), 482 (s). ESI(+) MS (m/z) MeCN: Calcd for [Fe(C29H20N6)(CH3CN)(CH3OH)]+ 545.1621, found 545.0291.
Acknowledgements
This work was supported by grants from the University of Otago and the MacDiarmid Institute, including the award of a MacDiarmid-funded PhD scholarship, and subsequently the award of a University of Otago Publishing Bursary, to JO.References
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44 CrossRef CAS.
- J.-F. Letard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 234, 221 CrossRef.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1 Search PubMed.
- S. Brooker and J. A. Kitchen, Dalton Trans., 2009, 7331 Search PubMed.
- K. S. Murray and C. J. Kepert, Top. Curr. Chem., 2004, 233, 195 CAS.
- W. J. Stratton and D. H. Busch, J. Am. Chem. Soc., 1958, 80, 1286 CrossRef CAS.
- Y. Sunatsuki, R. Kawamoto, K. Fujita, H. Maruyama, T. Suzuki, H. Ishida, M. Kojima, S. Iijima and N. Matsumoto, Coord. Chem. Rev., 2010, 254, 1871–1881 CrossRef CAS.
- (a) N. Bréfuel, S. Shova, J. Lipkowski and J.-P. Tuchagues, Chem. Mater., 2006, 18, 5467 CrossRef; (b) N. Bréfuel, I. Vang, S. Shova, F. Dahan, J.-P. Costes and J.-P. Tuchagues, Polyhedron, 2007, 26, 1745 CrossRef; (c) N. Bréfuel, S. Shova and J.-P. Tuchagues, Eur. J. Inorg. Chem., 2007, 4326 CrossRef.
- A. Noble, J. Olguín, R. Clérac and S. Brooker, Inorg. Chem., 2010, 49, 4560 CrossRef CAS.
- J. Olguín and S. Brooker, New J. Chem., 2011, 35, 1242 RSC.
- (a) J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203 CrossRef; (b) M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS; (c) J. Klingele, S. Dechert and F. Meyer, Coord. Chem. Rev., 2009, 253, 2698 CrossRef CAS.
- G. B. Kauffman, R. A. Albers and F. L. Harlan, in Inorganic Syntheses, ed. R. W. Parry, 1970 Search PubMed.
- (a) C. R. Johnson, W. W. Henderson and R. E. Shepherd, Inorg. Chem., 1984, 23, 2754 CrossRef CAS; (b) J. A. Winter, D. Caruso and R. E. Shepherd, Inorg. Chem., 1988, 27, 1086 CrossRef CAS.
- Examples of N4O2iron(II) SCO complexes are also known, for example (a) L. Salmon, A. Bousseksou, B. Donnadieu and J.-P. Tuchagues, Inorg. Chem., 2005, 44, 1763 CrossRef CAS; (b) B. Weber, Coord. Chem. Rev., 2009, 253, 2432 CrossRef CAS; (c) G.-C. Xu, L. Zhang, H.-B. Xu, T. Zhang, Z.-M. Wang, M. Yuana and S. Gao, Chem. Commun., 2010, 46, 2554–2556 RSC.
- Y. Sunatsuki, R. Kawamoto, K. Fujita, H. Maruyama, T. Suzuki, H. Ishida, M. Kojima, S. Iijima and N. Matsumoto, Inorg. Chem., 2009, 48, 8784 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of crystal structure for complex [FeII(H2L1)2](BF4)2·¼(Et2O)·¼(H2O)·MeOH] and Mössbauer spectra. CCDC reference number 796801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00190f |
| This journal is © The Royal Society of Chemistry 2011 |