Evaluation of a symmetry-based strategy for assembling protein complexes†
Dustin P.
Patterson
a,
Ankur M.
Desai
ab,
Mark M. Banaszak
Holl
ab and
E. Neil G.
Marsh
*abc
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: nmarsh@umich.edu
bMichigan Nanotechnology Institute for Medicine and Biological Sciences, University of Michigan, Ann Arbor, MI 48109, USA
cDepartment of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA
First published on 20th September 2011
Abstract
We evaluate a strategy for assembling proteins into large cage-like structures, based on the symmetry associated with the native protein's quaternary structure. Using a trimeric protein, KDPG aldolase, as a building block, two fusion proteins were designed that could assemble together upon mixing. The fusion proteins, designated A-(+) and A-(−), comprise the aldolase domain, a short, flexible spacer sequence, and a sequence designed to form a heterodimeric antiparallel coiled-coil between A-(+) and A-(−). The flexible spacer is included to minimize constraints on the ability of the fusion proteins to assemble into larger structures. On incubating together, A-(+) and A-(−) assembled into a mixture of complexes that were analyzed by size exclusion chromatography coupled to multi-angle laser light scattering, analytical ultracentrifugation, transmission electron microscopy and atomic force microscopy. Our analysis indicates that, despite the inherent flexibility of the assembly strategy, the proteins assemble into a limited number of globular structures. Dimeric and tetrameric complexes of A-(+) and A-(−) predominate, with some evidence for the formation of larger assemblies; e.g. octameric A-(+) : A-(−) complexes.
Introduction
In this paper we evaluate a strategy for directing the assembly of proteins into cage-like structures. The strategy exploits the symmetry arising from the quaternary structure of the protein and a pair of complementary coiled-coil sequences that are designed to form an antiparallel heterodimeric coiled-coil. By genetically engineering two protein constructs with complementary coiled-coil sequences fused to their C-termini, we create a system that is capable of self-assembly into multimeric protein complexes. This strategy is illustrated in Fig. 1.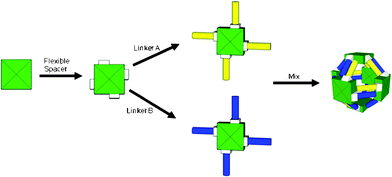 | ||
| Fig. 1 A strategy for self-assembly of protein cages based on the symmetry imparted by the quaternary structure of the protein. In this case the assembly of a cubic cage based on a tetrameric protein is illustrated. | ||
The assembly of individual protein subunits into higher order (quaternary) structures is essential for the biological function of many proteins. The design of proteins that self-assemble into well-defined, higher-order structures is an important goal that has applications in synthetic biology and in material science.1–9 In both natural and man-made protein assemblies, new properties emerge that are not manifested in the individual protein building blocks but arise from the assembly as a whole.
In Nature complex biological structures are often assembled from repeating units of only one or two protein building blocks and their assembly is strongly guided by symmetry. Multimeric protein assemblies may be broadly classified as either extended (filamentous) or closed (cage-like) structures: examples of the former include collagen, fibrin, actin and tubulin,10–13 whereas the second group is represented by icosahedral virus capsid proteins,14–15 ferritins,16 the pyruvate dehydrogenase core,17 the GroEL/GroES chaperonin complex18–19 and the proteosome complex.20 In each case, the quaternary structure is integral to the biological function of the protein: thus the fibers formed by collagen reflect that protein's structural role in connective tissue; the assembly of proteins into a capsid is required to package and protect the viruses' genetic material; the hollow barrel-like structure adopted by the proteosome subunits sequesters the protease active sites from the cellular milieu and prevents degradation of undamaged proteins.
The remarkable diversity of structural and functional properties exhibited by proteins suggests that assembling them into new quaternary structures would be a promising avenue for the construction of novel, responsive biomaterials.21–22 For example, the conditions for assembly could be made dependent on general properties that influence protein:protein interactions such as pH, ionic strength and redox potential, initiated by binding metal ions or other ligands, or by a specific enzyme-catalyzed modification of the protein.
There are numerous examples in which proteins and peptides have been assembled through various strategies into extended structures as fibers, gels or two-dimensional networks.4,23–28 Such structures lend themselves to the design of materials with physical properties that are responsive to stimuli such as those mentioned above. However, we have focused on the de novo assembly of cage-like structures, for which there are far fewer examples.29–30 These could be useful for the encapsulation, delivery and release of therapeutic agents, or the construction of multi-enzyme complexes.
Results
Design strategy
Our strategy for assembling proteins comprises two components: a protein “building block”, with well-defined quaternary structure, and two short “linker” sequences designed to dimerize with each other. To test our design strategy we chose KDPG-aldolase from T. maritima31 as a building block, for which an over-expressing clone was available. The crystal structure of the protein32 shows it to be a C3 symmetric trimer (Fig. 2) that lends itself to the assembly of polyhedral structures based on triangles. The N- and C-termini of the protein are not folded so that grafting linker sequences to facilitate assembly should not perturb the structure. The enzyme has the advantages of being thermostable and easily purified. Aldolase activity can be conveniently assayed, which provides a check that the protein is correctly folded.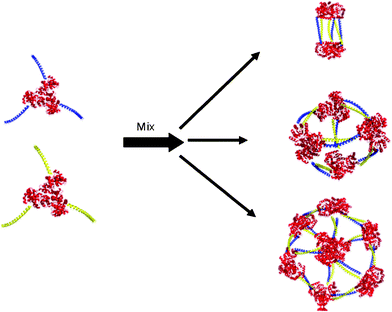 | ||
| Fig. 2 Cage structures that could be formed by the assembly of KDPG aldolase trimers (red) equipped with complementary coiled-coil linker domains (blue and yellow). | ||
As linkers we designed two complementary sequences intended to adopt an antiparallel heterodimeric coiled-coil structure upon dimerization (Fig. 3). The coiled-coil motif is one of the best understood protein–protein interactions.33–34 Coiled-coils can be designed to be parallel or antiparallel, and either homodimeric or heterodimeric, furthermore the strength of the interaction between the coils can be modulated by varying the length of the peptides.35 Because they are short, they can easily be grafted onto the N- or C-termini of other proteins and do not generally interfere with folding or activity.
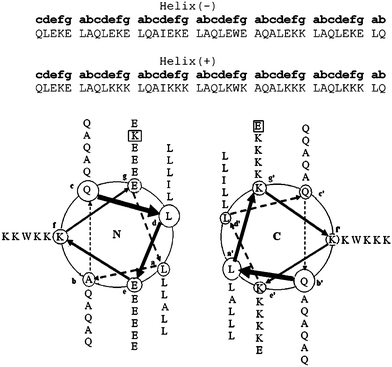 | ||
| Fig. 3 Design of hetero-dimeric antiparallel coiled-coil linker domains. Each domain was designed to encompass six heptad repeats with an antiparallel orientation being enforced by a “knobs-into-holes” Ile-Ala packing in the hydrophobic core and electrostatic interactions between the ‘e’-‘e`’ and ‘g’-‘g`’ interfaces. | ||
The coiled-coil linker domains were based on the antiparallel designs developed by Oakley and Hodges.36–38 We considered an antiparallel orientation to be desirable because this should minimize steric interactions between the larger aldolase trimers. By using a heterodimeric design we aimed to exert some control over the assembly process because no higher order structures should form until proteins equipped with complementary coiled-coil sequences are mixed. As shown in Fig. 3, the coiled-coil linkers were designed to comprise 6 heptad repeats. The hetero-dimeric interaction was established by complementary electrostatic interactions between residues at the interfacial ‘e’ and ‘g’ positions. The antiparallel orientation was enforced by incorporating complementary “knobs-into-holes” packing of Ala and Ile residues at the hydrophobic ‘a’ and ‘d’ positions that could only occur in the antiparallel orientation.
Each coiled-coil-forming sequence was introduced at the C-terminus of KDPG aldolase followed by a His6 tag sequence to facilitate purification. Since one helical sequence is predominantly positively charged and the other negatively charged, we refer to these proteins as A-(+) and A-(−). Both proteins were expressed in E. coli as soluble proteins, although at lower levels than the wild-type enzyme. The purified proteins could be concentrated to ≥10 mg/ml without precipitation occurring and were stable for several days at 4 °C.
To confirm that the helical linkers did not result in miss-folding of the protein, the activity of the fusion proteins was measured. kcat for the unmodified KDPG aldolase was found to be 2.6 ± 0.3 s−1 where kcat for A-(+) and A-(−) were 2.3 ± 0.2 s−1 and 2.2 ± 0.2 s−1 respectively. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of A-(+) and A-(−) exhibited similar activity, kcat = 2.3 ± 0.2 s−1, indicating that the addition of the helical sequence and does not significantly perturb the structure of the aldolase domain.
:1 mixture of A-(+) and A-(−) exhibited similar activity, kcat = 2.3 ± 0.2 s−1, indicating that the addition of the helical sequence and does not significantly perturb the structure of the aldolase domain.
We have also characterized the properties of the individual helical linkers (for details see supporting information), which could be expressed independently in E. coli. The isolated Helix-(+) was as expected unstructured in solution, as judged by its C.D. spectrum. Surprisingly, isolated Helix-(−) exhibited a C.D. spectrum characteristic of a highly α-helical peptide and appears to be a dimer as determined by sedimentation equilibrium ultracentrifugation, which suggests that it forms a homo-dimeric coiled-coil. Although the isolated Helix-(−) was not intended to be dimeric, when attached to the aldolase subunit we saw no evidence for self-dimerization of A-(−), as described below. Therefore in the context of our design strategy, Helix-(−) behaved as intended.
Self-assembly of A-(+) with A-(−)
To examine the oligomerization states of A-(+) and A-(−), and their complexes four complementary techniques were employed: size exclusion chromatography coupled with multi-angle laser light scattering (SEC-MALLS); transmission electron microscopy (TEM); analytical ultracentrifugation (AUC); and atomic force microscopy (AFM). In each case, the properties of the individual proteins were compared with the properties of the mixture.Size exclusion chromatography–multi-angle laser light scattering analysis
A chromatography system equipped with in-line refractive index (RI) and MALLS detectors was used which allowed real-time information on size, shape and the molecular weight distribution for the eluting protein species to be obtained. Fig. 4 shows the chromatograms for samples of A-(+) and A-(−). Both proteins eluted as single peak and appear monodisperse: the polydispersity indices of A-(+) and A-(−) were 1.01 and 1.02 respectively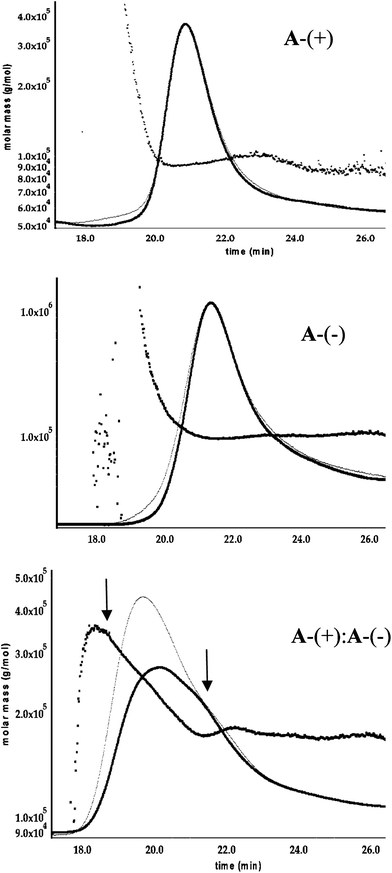 | ||
| Fig. 4 Analysis of A-(+) : A-(−) complex formation by SEC—MALLS. Detection is by refractive index (thin trace) and by MALLS (thick line). In all cases some tailing of proteins is observed. The number average molecular weight, Mn, of the eluted protein, calculated from MALLS, is shown plotted as a function of elution volume. A-(+) and A-(−) elute as monodisperse species as evidenced by the close correspondence of the MALLS and RI traces. The 1:1 mixture of A-(+) and A-(−) (bottom trace) clearly shows the presence of more than one species. Mn at the leading and lagging edges of the peak (marked by arrows) are consistent with the formation of a A-(+)2A-(−)2 tetramer and a A-(+)A-(−) dimer respectively. | ||
The number average molar masses, Mn, for A-(+) and A-(−) (calculated from the MALLS data) were 93.9 kDa and 102 kDa respectively. These values agree well with the calculated molecular weights of ∼90 kDa for the two fusion proteins. The rms radii of A-(+) and A-(−), determined by light scattering, are 7.2 nm and 6.8 nm respectively. These radii are larger than that of the un-modified aldolase (rms radius 5.5 nm) and reflect the expect increase in size due to the addition of the linker sequences to the fusion proteins.
Having established that the fusion proteins were well behaved, we investigated the ability of A-(+) and A-(−) to assemble into larger structures. 1:1 mixtures of A-(+) and A-(−) were incubated together for 2 h at 4 °C and their oligomerization state analyzed by size exclusion chromatography with MALLS/RI detection. The chromatograph for the mixture (Fig. 4) indicates that the samples are a mixture of species with decreased retention times, indicating formation of higher order oligomeric structures by A-(+) and A-(−).
A powerful advantage of MALLS/RI detection is that it allows the number-averaged molecular weight of eluting species to be analyzed as a function of peak cross-section and thus information on the species present can be obtained even if they are not cleanly resolved by the column. The molecular weight distribution plot of the chromatograph corresponding to the 2 h incubation indicates at least two major species are present. At the leading edge of the peak the eluting protein species is characterized by Mn ∼360 kDa and an rms radius of 12.3 ± 0.3 nm, whereas at the lagging edge Mn is ∼180 kDa and rms radius of 7 ± 1 nm. These molecular weights would correspond to the formation of an A-(+)2A-(−)2 hetero-tetramer and an A-(+)A-(−) hetero-dimer in the mixture. (To simplify nomenclature we use A-(+) and A-(−) to refer to the trimeric proteins, thus an A-(+)A-(−) hetero-dimer is understood to comprise a dimer formed between the two A-(+) and A-(−) trimers.) The decrease in Mn in between the two plateau regions is best accounted for by the overlap of two peaks of comprising the 360 kDa and 180 kDa species.
Analytical ultracentrifugation
Velocity sedimentation ultracentrifugation was used to analyze the sedimentation of the individual proteins and mixtures of A-(+) and A-(−). The sedimentation coefficient, s, depends upon both the molecular weight and frictional ratio, f/fo, of the sedimenting species and thus can be used to gain information on the both the size and shape of proteins. Both the individual A-(+) and A-(−) proteins and the complexes formed upon mixing A-(+) and A-(−) were stable under the centrifugation conditions. In particular, the sedimentation properties of the mixture appeared unchanged when the sample concentration was varied over a five-fold range. This suggests that the protein complexes in the mixture are not in dynamic equilibrium with each other. Also important, no loss of sample intensity was observed during centrifugation, indicating that large-scale protein aggregates (which would immediately sediment to the bottom of the cell) are not formed by A-(+) and A-(−).The sedimentation traces were subjected to enhanced van Holde-Weischet analysis39 to examine the distribution of sedimentation coefficients in each sample (Fig. 5). The unmodified aldolase trimer is extremely homogenous and is characterized by s20w = 4.6 S. The introduction of the coiled-coil domains introduces slight heterogeneity into A-(+) and A-(−), and the median s20w values for A-(+) and A-(−) increase slightly to 5.2 S and 5.8 S respectively. The observed heterogeneity may result from the inherent mobility of the linker sequences, although some self-association of the A-(+) and A-(−) trimers is also a possibility.
![Van Holde-Weischet [G(s)] plots of sedimentation coefficient distributions for sedimenting proteins calculated from sedimentation velocity ultracentrifugation data for the parent KDPG-aldolase (red circles); A-(+) (purple diamonds); A-(−) (green squares) and the 1 : 1 mixture of A-(+) : A-(−) (blue triangles).](/image/article/2011/RA/c1ra00282a/c1ra00282a-f5.gif) | ||
| Fig. 5 Van Holde-Weischet [G(s)] plots of sedimentation coefficient distributions for sedimenting proteins calculated from sedimentation velocity ultracentrifugation data for the parent KDPG-aldolase (red circles); A-(+) (purple diamonds); A-(−) (green squares) and the 1:1 mixture of A-(+) : A-(−) (blue triangles). | ||
As expected, the 1:1 mixture of A-(+) and A-(−) is more heterogeneous and the van Holde-Weischet plot is characterized by wider range of sedimentation coefficients that are larger than those for either individual protein (Fig. 5). The median s20w = 10 S for the mixture is close to that expected for an A-(+)A-(−) hetero-dimer. There is also a significant concentration of species with higher sedimentation coefficients ranging up to ∼25 S, indicating that higher order oligomers are formed. Assuming that these species are roughly spherical, the sedimentation coefficient distribution spans the range expected for an A-(+)2A-(−)2 hetero-tetramer. At the high end, the plot also indicates that even larger protein complexes, possibly as large as octamers, are present. Overall, the ultracentrifugation data are consistent with the results obtained by SEC-MALLS.
Transmission electron microscopy
TEM was used to visualize the complexes formed by A-(+) and A-(−). The protein complexes were first subjected to SEC on a sepharose S400 column that had been calibrated with standard proteins to provide partial separation of the complexes based on size. Fractions from the column were collected and analyzed by negative stain TEM. Typical images from two fractions are shown in Fig. 6. One fraction, Ve/Vo = 1.75–1.90, was expected to contain species with Mr in the approximate range 106–5 × 105 Da, the other (Ve/Vo = 2.05–2.20) was expected to contain species with Mr in the approximate range 5 × 105–105 Da. | ||
| Fig. 6 TEM images of A-(+) : A-(−) complexes after size exclusion chromatography. Left: representative field of view for proteins complexes eluting with apparent Mr ∼106–5 × 105 Da. Right: representative field of view for proteins eluting with apparent Mr ∼5 × 105–105 Da. Representative structures corresponding to individual A-(+) or A-(−) trimers are circled; structures with morphologies consistent with “collapsed” dimeric, tetrameric and octameric complexes are indicated by rectangles, triangles and squares. Samples were prepared by negative staining with uranyl formate. | ||
The individual subunits of the A-(+) and A-(−) trimers were resolved in the TEM image. The trimers are predominantly associated into discrete complexes with diameters of ∼10–20 nm. There was no evidence in any images for extended or fibrous structures. A range of particle sizes are evident, which is consistent with the results from size exclusion chromatography and analytical centrifugation. The process of depositing proteins on the grid and fixing the sample collapsed the open, cage-like structures that the A-(+) : A-(−) complexes are intended to adopt, so the 3-dimensional structures of the complexes cannot be inferred from the image. However, in a significant number of cases the number of A-(+)/A-(−) building blocks associated with the complex can be discerned; in particular, examples of complexes with two, four or higher numbers of building blocks are evident.
Atomic force microscopy
As a complementary form of molecular imaging, AFM was used to characterize the assembly of A-(+) and A-(−). Although the length scales of the proteins are too small for this technique to provide meaningful information in the x and y dimensions, AFM is very sensitive in the z dimension and was used to measure the height of the proteins. Depending upon the orientation of the aldolase trimer to the surface, height of the protein should be in the range of 3 to 5 nm. Individual samples of un-modified aldolase, A-(+) and A-(−) were imaged and the height distribution of particles analyzed (Fig. 7). The average height of particles in each sample was 3.5–4.0 nm, with essentially no particles larger than 9.5 nm. Samples of 1:1 mixtures of A-(+) and A-(−), however, showed a small but distinct sub-population of particles with heights of 8–12 nm, which would be consistent with the formation of larger structures.
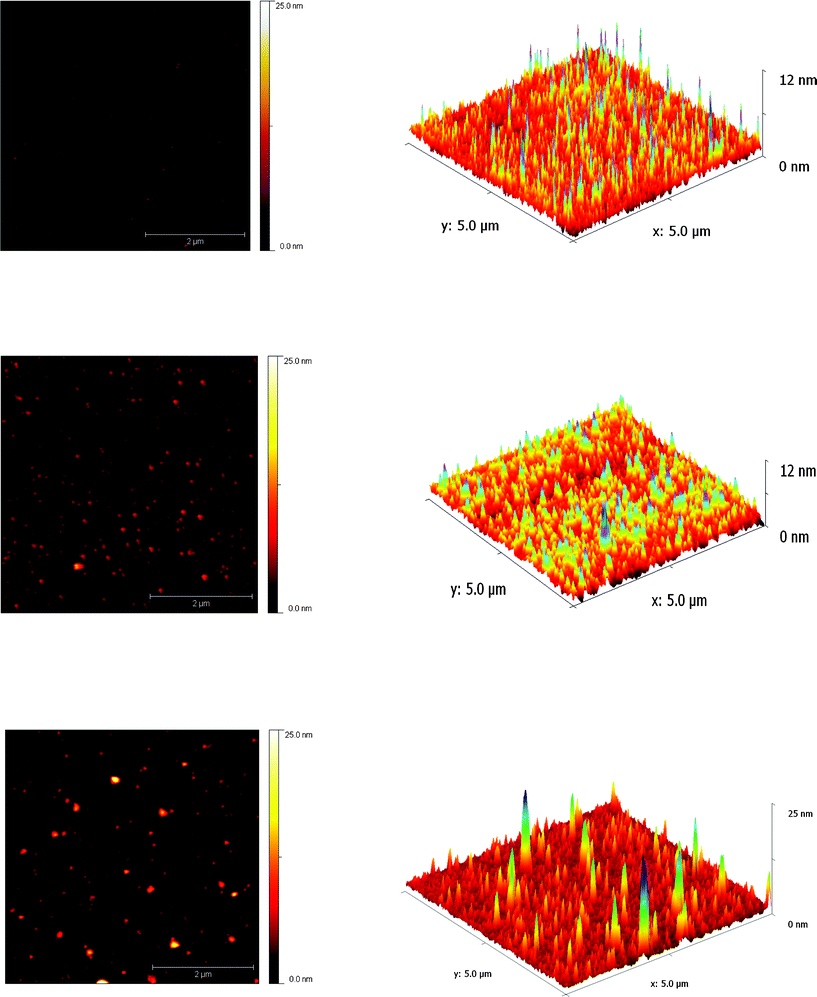 | ||
| Fig. 7
Atomic force microscopy of proteins. Left 2-D plots of surface height; Right 3-D reconstruction of the same data. Top image of A-(−) adsorbed on mica; middle image of A-(−) adsorbed on mica; bottom image of a 1:1 mixture of A(−) and A(+)adsorbed on mica. The scale bar in the AFM images represents 2 μm for all images. Differences in the x–y dimensions of particles are tip-induced artifacts. | ||
It is unclear why AFM did not reveal more convincing evidence for the assembly of A-(+) and A-(−), since the AUC, TEM and MALLS data presented above clearly support the formation of multimeric structures. Our failure to observe larger protein complexes may reflect the method used to prepare samples for AFM, which involves using dilute solutions and extensive washing of the mica sheets. We observed that the unmodified aldolase protein adhered poorly to the mica surface, making it hard to image, whereas the modified A-(+) and A-(−) proteins adhere well and were readily imaged. This suggests that the individual coiled-coil sequences interact strongly with the surface, which is presumably not possible when they are mediating complex formation. Thus the complexes may adhere only weakly, and so are either removed during dilution and washing, or dissociate to individual A-(+) and A-(−) components.
Discussion
In contrast to the assembly of DNA nano-structures, the design of structurally well-defined protein complexes has proved much harder to achieve. Previous studies have taken a “directed” approach towards the problem of assembling proteins into larger structures. For example, based on detailed analysis of X-ray crystal structures Yeates and coworkers designed a fusion protein in which two protein domains were precisely and rigidly oriented in the correct geometry for assembly into a tetrahedral cage.29 However, this approach requires identifying specific proteins that are compatible with the intended design and thus lacks generality.Therefore, in our studies we wanted to test a fundamentally different approach to protein assembly that could be applied to a wide range of proteins; in the present case any protein that possesses a homo-trimeric quaternary structure. By introducing some flexibility between the aldolase building block and the coiled-coil linker domain we aimed to allow the components sufficient freedom to assemble into structures that are compatible with the symmetry inherent to the quaternary structure of the protein. Thus, rather than forcing a specific structure on the protein, our study asks the question: given the ability to assemble into higher-order structures, what structure(s) will form?
An important design criterion for creating protein assemblies is that it should be generally applicable to many proteins. The use of coiled coils as a minimal structural unit to link larger proteins together appears to fulfil this criterion well. The coiled-coil interaction is robust and easily modulated through electrostatic interactions; moreover, because they are genetically encoded protein complexes could, if desired, be assembled in vivo. The aldolase fusion proteins retained the same activity as the unmodified enzyme and the isolated proteins remained as trimers, as judged by SEC and AUC. Furthermore, using a heterodimeric coiled-coil pair affords a measure of control over protein assembly because neither A-(+) nor A-(−) form higher-order oligomers until they are mixed together.
There are many structures that could, in principle, form by oligomerization of the triangular building block represented by the KDPG aldolase trimer. A back-to-back dimer of A-(+) and A-(−) represents the simplest complex and tetramers, octamers and 20-mers (representing tetrahedral, octahedral and icosahedral complexes respectively) are the most highly symmetrical structures that could form. However, any closed structure comprising an equal number of A-(+) and A-(−) trimers would satisfy the valency rules imposed by the need for each positive helix to associate with a negative helix. Extended 1-D and 2-D networks of aldolase trimers could also potentially form. We conjectured that although many structures may, in principle, be compatible with our simple design rules, in practice relatively few will be stable, and that symmetrical structures that can form “closed shell” complexes would be favored.
We used several different experimental techniques to characterize the assemblies produced by mixing the A-(+) and A-(−) proteins. The different techniques require different methods of sample preparation, which may influence distribution of structures formed, however SEC-MALLS, TEM and AUC each provided data that were consistent with A-(+) and A-(−) forming relatively few structures, rather than a mixture of all conceivable structures. The exception was of AFM that provided less conclusive evidence for the formation of protein complexes, which, as noted, above may be due to the sample adsorption characteristics.
The most prevalent structure, for which there is good evidence from SEC-MALLS and AUC, appears to be a dimer of trimers that would form by the back-to-back association of one A-(+) and one A-(−) trimer. This is clearly the simplest structure that could form, and indeed it would be surprising if it were not observed. More interesting is that more complex structures are formed in significant amounts. In particular, the SEC-MALLS and AUC data are consistent with the assembly of a an A-(+)2A-(−)2 tetrahedral cage. AUC and TEM also point to the formation of some larger species as minor components. Formation of these larger structures would be expected to be entropically unfavorable with respect to forming a simple A-(+)A-(−) dimer. However, if forming a dimer imposes an unfavorable conformation on the protein that is relieved upon opening up of the structure to form tetramers or octamers, this would favor formation of these larger complexes.
We note that a tetrahedrally symmetrical A-(+)2A-(−)2 tetramer cannot form if the A-(+) and A-(−) trimers remain associated as homo-trimers because the correct coiled-coil interactions cannot form. However, if the individual A-(+) and A-(−) subunits equilibrate between trimers, so that hetero-trimers (comprising one A-(+) and two A-(−) subunits, or vice versa) are formed, then a tetrahedron can readily be constructed (Fig. 2). The “shuffling” of subunits between trimer, although hard to detect, may reasonably be expected to occur since they are non-covalently associated, and the monomeric form of the enzyme has been produced by a simple point mutation of the trimer interface.31
The extent to which the mixture of complexes formed is governed by kinetic or thermodynamic factors is currently unclear. Varying the speed of mixing did not seem to significantly alter the distribution of complexes formed, which might suggest that the distribution represents a thermodynamic mixture. However, ultracentrifugation experiments, as described above, found no change in the sedimentation profile of the complexes over a 5-fold range of concentrations. This result suggests the complexes are not in dynamic equilibrium and may be kinetically trapped. Attempts to thermally unfold and re-anneal the mixture of complexes were unsuccessful as aggregation and precipitation occurred on cooling (D.P.P. unpublished observations).
Ideally, any strategy for engineering self-assembling protein cages would aim for the formation of a single, well-defined structure. Although we have not achieved this aim here, we note that these studies represent only the first iteration of the design. We believe there is considerable scope for optimizing the system to adopt one of the limited number of structures that appear to be energetically favorable. Optimization of the coiled-coil interaction by, for example, altering the strength, length or orientation (parallel vs. antiparallel) of the coiled coils, and/or adjusting the assembly conditions, could be used to favor the formation of one complex over other complexes of similar kinetic or thermodynamic stabilities, resulting in a unique design solution.
Conclusions
We have shown that the attachment of short, de novo-designed coiled-coil-forming sequences to a natural protein results in a self-assembling system. The constraints on the possible ways in which the proteins could assemble were deliberately minimal, but it appears, perhaps counter-intuitively, that they form a relatively small number of complexes. These complexes were characterized by a variety of experimental techniques; their properties are consistent with them assembling into cage-like structures that are guided by the C3 symmetry associated with the protein's trimeric quaternary structure. The design strategy can in principle be extended to a wide variety of proteins with different quaternary structures.Experimental
Materials
DNA modifying enzymes and reagents were purchased from New England Biolabs. DNA primers were purchased from IDT DNA Technologies (Coralville, IA). PBS 10× stock solution was purchased from Invitrogen (Carlsbad, CA). E. coliBL21(λDE3) and expression vector pET-28b were purchased from Novagen (Madison, WI); Pfu turbo DNA polymerase and E. coli XL1-Blue were from Stratagene (Cedar Creak, TX). Nickel nitrilotriacetic acid (Ni-NTA) resin, QIAquick gel extraction kit, and QIAprep Spin Miniprep kit were purchased from Qiagen (Valencia, CA). DNP-10 non-condensed silicon nitride tips were purchased from Veeco Probes (Camarillo, CA). Gold nanoparticles were from Ted Pella, Inc. (Redding, CA). Vinlyec (polyvinyl formal) film grids, stabilized with carbon, were purchased from Ernest F. Fullam, Inc. (Clifton Park, NY). All the chemical reagents were from Fisher Scientific (Pittsburgh, PA).DNA constructs
The gene encoding T. maritimaKDPG Adolase31 in pUC19 was kindly provided by Carol Fierke and Manoj Cheriyan (Univ. of Michigan). Genes encoding the sequences of the coiled-coil linkers helix-(+) and helix-(−) were designed and were commercially synthesized and sub-cloned in pET28b (Picoscript, Houston, TX). The gene encoding KDPG aldolase was excised from pUC19 by digestion with Nco I and Xho I restriction enzymes. The fragment was purified and ligated into containing into pET28b-based constructs containing genes encoding helix-(+) and helix-(−), similarly digested with Nco I and Xho I, to achieve the desired gene fusion. Expression vectors containing the recombinant gene fusions were transformed into E. coliBL21(λDE3) cells to facilitate protein expression using standard procedures.Protein expression and purification
E. coli strains harboring expression constructs were grown on 2×TY medium at 37 °C in the presence of kanamycin to maintain selection for the plasmids. Expression of the genes were induced by addition of isopropylβ-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM once the cells reached early log phase (OD600 = 0.8). Cultures were grown for 4 h after addition of IPTG, then the cells were harvested by centrifugation and cell pellets stored at −20 °C until needed.Cell pellets were resuspended in ice-cold Lysis buffer (50 mM Tris-HCl, 150 mM sodium chloride, 10% glycerol, pH 8.0) containing 1 mM β-mercaptoethanol, 0.1 mM PMSF and lysed by sonication on ice. Cell debris was removed by centrifugation at 24,000 g for 15 min at 4 °C. The supernatant was decanted into another centrifuge tube and heated at 80 °C in a water bath for 30 min to produce a white cloudy precipitate. Precipitated material was removed by centrifugation at 24,000 g for 15 min at 4 °C. The supernatant was loaded onto a 5 mL column of Ni-NTA superflow resin equilibrated in buffer A (50 mM Tris-HCL, 300 mM NaCl, 2.5 mM imidazole, 1 mM β-mercaptoethanol, 10% glycerol, pH 8.0) at a flow rate of 0.5 mL/min using a Biologic HR FPLC (Biorad, CA)and the column washed with 30 mL of buffer A. Non-specifically bound proteins were eluted with a step gradient (20 mL at flow rate of 2 mL/min for each step) of increasing concentrations (7.5 mM, 27.5 mM and 52.5 mM) of imidazole in buffer A. Finally the His6-tagged fusion proteins were eluted with buffer A containing 500 mM imidazole. This yielded protein that was greater than 95% pure as judged by SDS-PAGE. Purified protein solutions were dialyzed twice overnight against 1.5 L storage buffer (50 mM Tris-HCL, 150 mM NaCl, 0.5 mM EDTA, 10% glycerol, pH 8.0) and stored at −20 °C. Protein concentrations were determined by UV absorption measured at 280 nm assuming a molar extinction coefficient ε280 = 12,900 M−1cm−1.
The fusion protein of aldolase with Helix-(+), A-(+), was found to bind nucleic acids tightly and so the purification procedure was modified slightly. To remove nucleic acids the protein was bound to Ni-NTA column as described above and then washed with 30 mL of denaturing buffer (50 mM, Tris, 6 M guanidium-HCl, 300 mM NaCl, 5 mM imidazole, 1 mM β-mercaptoethanol, 10% glycerol, pH 8.0). The protein was refolded on the column using a decreasing linear gradient of denaturing buffer with buffer A, (60 mL total volume, flow rate 0.5 mL/min) before being eluted with 500 mM imidazole.
Enzyme assay
Catalytic activity was assayed using the lactate dehydrogenase-coupled assay. HEPES (10 mM, pH 8.0, 100 μL), NADH (230–280 μM), lactate dehydrogenase (0.8 Units), KDPG (1 mM) were mixed in a quartz cuvette at 34 °C. The assay was started by addition of KDPG aldolase (final concentration 1 μM) or the fusion proteins (final concentrations 0.5–0.6 μM) and activity followed by monitoring the decrease in absorbance at 340 nm.Size exclusion chromatography of proteins
Protein samples were dialyzed against sodium phosphate buffer (1 mM, pH 8.0) in order to remove salts. Dialyzed protein solutions were then flash frozen with liquid nitrogen and lyophilized. Prior to injection, the protein samples were reconstituted in PBS buffer (1.0 mM potassium phosphate, 155 mM NaCl, 3.0 mM sodium phosphate, pH 7.4) and allowed to incubate at 4 °C for 2 h or 24 h before injection. 100 μL of each sample, 1–2 mg/mL was used for each chromatographic analysis. Samples of commercially available bovine serum albumin (BSA) and carbonic anhydrase (CA) were used as standards.GPC experiments were performed on an Alliance Waters 2695 separation module equipped with a 2487 dual wavelength UV absorbance detector (Waters Corporation), a Wyatt HELEOS Multi Angle Laser Light Scattering (MALLS) detector, and an Optilab rEX differential refractometer (Wyatt Technology Corporation). Three columns connected in series were employed to separate samples TosoHaas TSK-Gel G 2000 PW 05761 (300 mm × 7.5 mm), G 3000 PW 05762 (300 mm × 7.5 mm), and G 4000 PW (300 mm × 7.5 mm). The column temperature was maintained at 25 ± 0.1 °C with a Waters temperature control module. The columns were equilibrated in PBS pH 7.4, and samples eluted at 1 mL/min. The number average molecular weight, Mn, was calculated from the MALLS and differential refractive index data using Astra 5.3.14 software (Wyatt Technology Corporation).
Atomic force microscopy of proteins
Mica Sheets, 1 × 1 cm, attached to metal pucks were incubated with 40 μl HEPES buffer (10 mM, pH 7.0) at room temperature for 30 min. 0.5–1 μL of protein sample solutions (6 μM—100 μM concentrations) were applied to the center of the mica sheets and allowed to incubate for 30 min at room temperature. 250 μl of HEPES buffer was then applied to the mica surface to stop protein absorption. The mica was then washed with 4 mL of HEPES buffer to remove unbound protein and the sample imaged.Imaging was performed under tapping mode on a Nanoscope IIIa Multimode AFM (Digital Instruments, Veeco Metrology, Santa Barbra, CA) equipped with an “E” scanner. Veeco DNP-10 Non-Condensed Silicon Nitride tips (force constant = 0.06 N/m) were used for imaging under HEPES Buffer (10 mM, pH 7.0) filtered with a 0.2 μM filter. Typical scan sizes were 5 μm × 5 μm or 2 μm × 2 μm and were taken at 0.5–1 Hz scanning speeds. Images were processed and height distribution determined using the program Gwyddion.
Transmission electron microscopy of proteins
10 μL samples of proteins, 6 μM, were applied to form coated grids and incubated for 1–2 min and excess liquid was wicked away with filter paper. Grids were then washed twice with 10 μl of distilled water, wicking away liquid after each addition with filter paper, and then stained with 1 μl 1% uranyl formate for 1 min and excess stain was wicked away with filter paper. Images were obtained at room temperature at an accelerating voltage of 100 kV on a Morgagni 268(D) transmission electron microscope equipped with a Orius SC200W CCD camera.Analytical ultracentrifugation
Sedimentation velocity analysis was performed using a Beckman analytical ultracentrifuge equipped with an AN50TI rotor. Samples were dialyzed against PBS buffer, pH 8.0, containing 5% glycerol. Immediately prior to centrifugation, samples were filtered through 0.1 micron ‘anatop’ filters (Whatmann). The hydrodynamic behavior of A-(−) and A-(+) was analyzed at several different protein concentrations with initial absorptions of 0.2, 0.4, 0.6, 0.8, and 1.0 at 280 nm. Samples containing a 1:1 molar ratio of A-(−) and A-(+) were incubated overnight at 4 °C prior to filtering and centrifugation. Samples were loaded into sector-shaped double channel centerpieces, and allowed to equilibrate at 25 °C for 2 h in the non-spinning rotor prior to sedimentation. The individual A-(−) and A-(+) proteins were sedimented at 25,000 rpm whereas the mixture was analyzed at both 14,000 and 35,000 rpm to assist in the analysis of multiple species formed. Absorbance data were collected at a wavelength of 280 nm. Sedimentation velocity data were analyzed using the enhanced van Holde-Weischet analysis.39
Acknowledgements
We thank Dr Min Su and Justin Schilling for help with TEM and Dr Titus Franzmann for advice and help with the analytical ultracentrifugation experiments. D.P.P. acknowledges the support of NIH funded Chemistry Biology Interface training grant T32 GM008597. The authors gratefully acknowledge the use of MNIMBS instrumentation facility for SEC-MALLS analysis of proteins.References
- D. Papapostolou and S. Howorka, Mol. BioSyst., 2009, 5, 723–732 RSC.
- K. Channon, E. H. C. Bromley and D. N. Woolfson, Curr. Opin. Struct. Biol., 2008, 18, 491–498 CrossRef CAS.
- K. Sugimoto, S. Kanamaru, K. Iwasaki, F. Arisaka and I. Yamashita, Angew. Chem., Int. Ed., 2006, 45, 2725–2728 CrossRef CAS.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS.
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464–470 CrossRef CAS.
- T. Douglas and M. Young, Nature, 1998, 393, 152–155 CrossRef CAS.
- Z. Varpness, J. W. Peters, M. Young and T. Douglas, Nano Lett., 2005, 5, 2306–2309 CrossRef CAS.
- Z. Gu, A. Biswas, M. X. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- Z. Yang, X. Y. Wang, H. J. Diao, J. F. Zhang, H. Y. Li, H. Z. Sun and Z. J. Guo, Chem. Commun., 2007, 3453–3455 RSC.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS.
- A. S. Wolberg, Blood Rev., 2007, 21, 131–142 CrossRef CAS.
- E. Reisler and E. H. Egelman, J. Biol. Chem., 2007, 282, 36133–36137 CrossRef CAS.
- E. Mandelkow and E. M. Mandelkow, Curr. Opin. Cell Biol., 1989, 1, 5–9 CrossRef CAS.
- R. J. Kuhn and M. G. Rossmann, Adv. Virus Res., 2005, 64, 263–284 CrossRef CAS.
- M. G. Rossmann and J. E. Johnson, Annu. Rev. Biochem., 1989, 58, 533–573 CrossRef CAS.
- E. C. Theil, Annu. Rev. Biochem., 1987, 56, 289–315 CrossRef CAS.
- A. Mattevi, G. Obmolova, E. Schulze, K. H. Kalk, A. H. Westphal, A. Dekok and W. G. J. Hol, Science, 1992, 255, 1544–1550 CAS.
- P. B. Sigler, Z. H. Xu, H. S. Rye, S. G. Burston, W. A. Fenton and A. L. Horwich, Annu. Rev. Biochem., 1998, 67, 581–608 CrossRef CAS.
- K. A. Krishna, G. V. Rao and K. Rao, Curr. Protein Pept. Sci., 2007, 8, 418–425 CrossRef CAS.
- D. Y. Kim and K. K. Kim, J. Biol. Chem., 2003, 278, 50664–50670 CrossRef CAS.
- E. H. C. Bromley, K. Channon, E. Moutevelis and D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50 CrossRef CAS.
- K. Chockalingam, M. Blenner and S. Banta, Protein Eng., Des. Sel., 2007, 20, 155–161 CrossRef CAS.
- K. Usui, T. Maki, F. Ito, A. Suenaga, S. Kidoaki, M. Itoh, M. Taiji, T. Matsuda, Y. Hayashizaki and H. Suzuki, Protein Sci., 2009, 18, 960–969 CrossRef CAS.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS.
- A. J. Scotter, M. Guo, M. M. Tomczak, M. E. Daley, R. L. Campbell, R. J. Oko, D. A. Bateman, A. Chakrabartty, B. D. Sykes and P. L. Davies, BMC Struct. Biol., 2007, 7 Search PubMed.
- S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510–5514 CrossRef CAS.
- J. S. Elam, A. B. Taylor, R. Strange, S. Antonyuk, P. A. Doucette, J. A. Rodriguez, S. S. Hasnain, L. J. Hayward, J. S. Valentine, T. O. Yeates and P. J. Hart, Nat. Struct. Biol., 2003, 10, 461–467 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS.
- T. W. Ni and F. A. Tezcan, Angew. Chem., Int. Ed., 2010, 49, 7014–7018 CrossRef CAS.
- J. S. Griffiths, N. J. Wymer, E. Njolito, S. Niranjanakumari, C. A. Fierke and E. J. Toone, Bioorg. Med. Chem., 2002, 10, 545–550 CrossRef CAS.
- S. W. Fullerton, J. S. Griffiths, A. B. Merkel, M. Cheriyan, N. J. Wymer, M. J. Hutchins, C. A. Fierke, E. J. Toone and J. H. Naismith, Bioorg. Med. Chem., 2006, 14, 3002–3010 CrossRef CAS.
- E. Moutevelis and D. N. Woolfson, J. Mol. Biol., 2009, 385, 726–732 CrossRef CAS.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- J. Y. Su, R. S. Hodges and C. M. Kay, Biochemistry, 1994, 33, 15501–15510 CrossRef CAS.
- M. G. Oakley and J. J. Hollenbeck, Curr. Opin. Struct. Biol., 2001, 11, 450–457 CrossRef CAS.
- D. G. Gurnon, J. A. Whitaker and M. G. Oakley, J. Am. Chem. Soc., 2003, 125, 7518–7519 CrossRef CAS.
- O. D. Monera, N. E. Zhou, P. Lavigne, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1996, 271, 3995–4001 CrossRef CAS.
- B. Demeler and K. E. van Holde, Anal. Biochem., 2004, 335, 279–288 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00282a/ |
| This journal is © The Royal Society of Chemistry 2011 |