DOI:
10.1039/C1RA00357G
(Paper)
RSC Adv., 2011,
1, 1585-1592
Preparation of hydrogen and carbon nanotubes over cobalt-containing catalystsviacatalytic decomposition of ethanol
Received
25th June 2011
, Accepted 26th August 2011
First published on 26th October 2011
1. Introduction
Energy resources and the environment are basically two problems faced by us. There are several kinds of energy in our world, such as solar energy, wind energy, nuclear energy, water power, fossil source, biomass energy, hydrogen power and so on. Among them, hydrogen is considered as an attractive power source due to its high conversion efficiency and low pollutant emissions.1 It can be produced traditionally by water electrolysis, gasification reactions, partial oxidation reactions of heavy oil and hydrocarbon steam reforming reactions.2,3 Recently, chemical decomposition of biomass materials like natural gas, methanol and ethanol is another way to produce hydrogen.4–9 Among the various sources that can be converted into hydrogen, ethanol is a very promising candidate due to its low production costs, high hydrogen content and low toxicity. Extensive studies have been carried out to find an efficient, stable and cheap catalyst for the hydrogen production.7–9 And the successful production of hydrogen over various catalysts by catalytic decomposition of ethanol has been reported by many research groups. However, to our knowledge, there are few works reported the production of carbon nanotubes (CNTs) accompanied by hydrogen production during ethanol decomposition.
CNTs, a two-dimensional nanostructure of carbon materials, have attracted intense attention worldwide because of their excellent physical and chemical properties.10 Currently, chemical vapor deposition (CVD) is known as a simple and convenient method for mass production of CNTs.11 Although the successful synthesis of CNTs by CVD of ethanol or other method has been reported by many researchers,12–18 the production of hydrogen accompanied by CNT growth has not been systematically investigated in their research work.
Our previous work has reported the co-production of hydrogen and MWCNTs over Fe catalysts.19 The influence of several experimental parameters such as reaction temperature, reaction time and feeding rates of ethanol on the catalytic activity of ethanol decomposition has been systemically investigated. The results showed that 800 °C was the optimum temperature for catalytic decomposition of ethanol to produce hydrogen and MWCNTs when the feeding rate of ethanol was 0.3 ml h−1. However, it should be noted that the temperature of 800 °C is uneconomic for CVD reaction due to its high energy consumption. Recent studies have suggested Co as the most suitable metal for hydrocarbon decomposition to synthesize hydrogen or MWCNTs at relatively low temperature.20,21 In order to reduce energy consumption and obtain hydrogen and MWCNTs at low temperature (< 800 °C), herein, we try to design a series of Co/Al2O3 catalysts and investigated their catalytic activity on ethanol decomposition. The purpose of the work is to explore optimum experiment conditions to produce hydrogen and high-quality MWCNTs at relatively low temperature, which may have important technological application in the field of energy.
2. Experimental section
2.1 Preparation of catalysts
Co/Al2O3 catalysts with different loading amounts of Co were prepared using a hydrothermal method. The loading amount of Co in the Co/Al2O3 catalysts was adjusted to X mol% of total metal (Co and Al) cations (10 mol% means Co/(Co + Al) = 10%) and varied from 10 to 90 mol% at 10 intervals. The detailed procedure of the catalyst preparation is as follows: an aqueous solution of 10 ml containing predetermined amounts of Co(NO3)2·6H2O and Al(NO3)3·9H2O was first prepared. The pH value of the solution was adjusted to 8 by adding NH3·H2O solution of 25 wt.% gradually with constant stirring to obtain the catalysts with different loadings of Co. The mixture was aged at room temperature for 4 h. Thus, a binary colloid was achieved. The colloid was placed in a 25 ml capacity Teflon-lined stainless steel autoclave. It was heated at 140 °C for 48 h. The hydrothermal products were collected by sedimentation and washed with deionized water 5 times, followed by drying at 80 °C for 12 h. The prepared catalysts or catalyst precursors were denoted as Co (X mol%)/Al2O3, where X mol% is the mol% loading in terms of Co.
The reaction apparatus used for ethanol decomposition was a conventional gas flow system with a vertical fixed bed at the center of a quartz tubular reactor (inner diameter 5 mm). The temperature of the bed was monitored with a thermocouple attached to the outside wall of the reactor in close proximity to the catalyst disc. Prior to ethanol decomposition, the catalyst (150 mg) was reduced by hydrogen at 500 °C for 1 h and purged under Ar at the same temperature for 30 min. Then the decomposition of ethanol was performed on the Co/Al2O3 catalysts at the temperatures of 500, 600, 700 and 800 °C. Ethanol was injected through a microinjector into a vaporizing chamber (100 °C) at a flow rate of 0.3 ml h−1 and switched to the tubular reactor using Ar (40 ml min−1) as the carrier gas. During ethanol decomposition, the gaseous products in the exit gas from the tubular reactor were analyzed at 6 min intervals by a gas chromatograph with a flame ionization detector (GC/FID). After 50 min, the ethanol feed was stopped. The solid products were collected from the tubular reactor.
2.3 Characterization
Temperature programmed reduction with hydrogen (H2-TPR) was performed to determine the reduction behavior of the catalysts with different loading amounts. These experiments were carried out in an U-shaped tubular quartz reactor, using 50 mg of catalyst and a temperature ramp from 25 to 1000 °C of 10 °C min−1. A flow rate of 40 ml min−1 of 5% H2 in Ar was used. A thermal conductivity detector was employed to determine the amount of hydrogen consumed. The scanning electron microscopy (SEM) images of the catalysts with solid products by ethanol decomposition were measured using a FEI Quanta 400 ESEM-FEG (environmental scanning electron microscope-field emission gun) equipped with X-ray energy dispersive spectrometer (EDS). The transmission electron microscope (TEM) images of MWCNTs were recorded with a JEOL JEM-3010 high-resolution transmission electron microscope (HRTEM) at an accelerating voltage of 300 kV. X-Ray diffraction (XRD, D/max-3c, Cu Ka, 50 kV, 300 mA, at room temperature in air) was used to characterize the compositions of the catalyst after reduction with hydrogen at 500 °C and of the catalyst with solid products after ethanol decomposition.
3. Results and discussion
3.1 Characterization of the catalysts
The H2-TPR patterns of the 10, 30, 50, 70, 90 mol% Co supported on Al2O3 catalysts are plotted in Fig. 1. It is clearly evident from the patterns that with increasing in Co loadings the H2 uptakes are increased. All materials show a shoulder at 290 °C and a principal peak at 400 °C, which are attributed to the reduction of Co3O4 to CoO and subsequent reduction of CoO to metallic Co, respectively.22
 |
| | Fig. 1
H2-TPR profiles for the Co/Al2O3 catalysts. | |
Fig. 2 displays the XRD pattern of the reduced Co(30 mol%)/Al2O3 catalyst. The main crystallographic planes of fcc metallic cobalt are detected at 44.2 (111), 51.4 (200) and 75.8° (220) (JCPDS 15-0806). The character diffraction peaks at 2θ = 37.7 and 68.2° can be attributed to the crystal planes of γ -Al2O3.23 The above results show that only the peaks due to Co metal and Al2O3 can be observed in the XRD pattern, indicating the catalyst has been reduced at 500 °C. Fig. 3a shows the SEM image for the surface morphology of Co(30 mol%)/Al2O3 catalyst after hydrogen reduction at 500 °C. It can be seen that the Co metallic particles were formed uniformly on the Al2O3 substrate with only a few particles aggregated together. A typical EDS analysis of the catalyst shown in Fig. 3b reveals the presence of Co, Al and O, consistent with the XRD results.
The changes of absolute yield of H2 as a function of Co loadings in ethanol decomposition with different temperatures are given in Fig. 4. From it, the total amount of H2 produced in 50 min could be clearly observed. At 500 °C, the total amount of H2 changed a little with the increase of Co loadings, which maintained at ∼5500 μmol. As the temperature reached to 600 °C, the total amount of H2 was slightly increased and mostly kept in the range of 7000–8000 μmol. The maximum amount of H2 could be obtained as the temperature increased to 700 °C, ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μmol H2 produced over Co(50 mol%)/Al2O3 catalyst. However, when the reaction temperature reached to 800 °C, the total amount of H2 began to decrease, lower than the results obtained at 700 °C or even lower than that at 600 °C over 10–40 mol% loading catalysts.
000 μmol H2 produced over Co(50 mol%)/Al2O3 catalyst. However, when the reaction temperature reached to 800 °C, the total amount of H2 began to decrease, lower than the results obtained at 700 °C or even lower than that at 600 °C over 10–40 mol% loading catalysts.
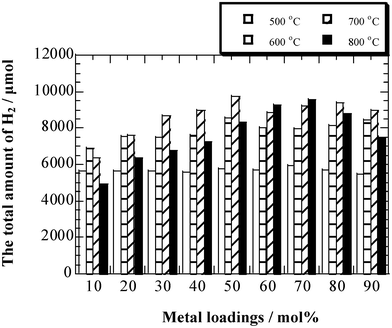 |
| | Fig. 4 The variation tendency of the absolute yield of H2 from ethanol decomposition in 50 min at the desired reaction temperature over Co/Al2O3 catalysts with different loading amounts. | |
We evaluated the catalytic performance of Co/Al2O3 catalysts to investigate the reason that lead to the variation of the total amount of H2 with temperatures. The result is shown in Fig. 5. It displays the changes of H2 yield as a function of the reaction time for ethanol decomposition over Co/Al2O3 catalysts at the temperatures of 500, 600, 700 and 800 °C. Here the H2 yield is calculated according to eqn (1).
| |  | (1) |
 |
| | Fig. 5 Changes of H2 yield as a function of the reaction time of 50 min in ethanol decomposition over Co/Al2O3 catalysts with different Co loadings at different temperatures: (a) 500, (b) 600, (c) 700, and (d) 800 °C. | |
From Fig. 5 it can be found that the H2 yield depends strongly on the amount of Co loadings and the reaction temperature. It is apparent from Fig. 5(a) that the H2 yield increased slightly (45–55%) as the Co loadings rise from 10 to 90 mol% at 500 °C. The result indicated that low temperature of 500 °C could not effectively enhance the catalytic activity of Co. A relatively great increase in the H2 yield is observed in Fig. 5(b) when the temperature reached to 600 °C, showing the catalysts with higher catalytic activities at 600 °C than those at 500 °C. As the temperature increased to 700 °C, the H2 yield decreased sharply on the catalysts with 10 and 20 mol% Co loadings, suggesting the deactivation of Co/Al2O3 catalysts occurred more easily on relatively lower Co loading catalysts. On the other hand, the catalytic activities over the catalysts with 30–90 mol% Co loadings enhanced slightly when compared with the result obtained at 600 °C. The maximum amount of H2 thus could be obtained at this temperature. As the reaction temperature reached 800 °C, the obvious deactivation of Co/Al2O3 occurred on each catalyst at the reaction time of 50 min. And it can be clearly seen that the lower the Co loadings, the faster the decrease of H2 yield. Previous studies of catalyst deactivation by carbon deposition have shown that the catalyst deactivation rate is a consequence of the imbalance between the carbon formation rate and the carbon diffusion rate.24,25 If the formation and diffusion rates were equal, active Co metal sites would be available and therefore the observed catalyst activity would remain stable. When the carbon diffusion rate through the catalyst particle was slower than that of the formation rate the active sites of catalyst would be covered or encapsulated by amorphous carbon and CNFs, which would deactivate the catalyst. From this viewpoint, it suggests that 800 °C should not be chosen for ethanol decomposition to synthesize hydrogen when using Co as catalyst.
In this section, a comparative study carried out by electron microscopy of the carbon deposited is presented. We mainly analyzed the carbon products synthesized over Co(30 mol%)/Al2O3 catalysts. Similar results can also be obtained over the catalysts with other loading amounts.
Fig. 6 shows the XRD pattern of solid products formed by ethanol decomposition over Co(30 mol%)/Al2O3 catalysts at 600 °C, from which a strong peak at 2θ = 26.1° representing the (002) graphitic basal plane reflection in the sample can be clearly seen. Although the peak is not suitable to differentiate structure details between CNTs and other graphite-like structures, it provides a primary evidence of graphite deposits.26 The XRD pattern of the reaction products also shows the peaks of metallic cobalt and Al2O3, which indicates that the initial structure of the catalysts did not undergo any substantial transition during the reaction.
SEM was carried out in order to observe the typical morphologies of the samples obtained. Fig. 7 shows the SEM images of deposited products formed by ethanol decomposition over Co(30 mol%)/Al2O3 catalyst at different temperatures. The distributions of external diameters of tubes or fibers are inserted in it. The four images of carbon products in Fig. 7(a)–(d) show nearly the same features. Web-like, micrometre long nanotubes or nanofibers were synthesized successfully and entangled together at each temperature. However, there also exists some differences among these samples due to the gradually increase of reaction temperature, which mainly influence the diameter distribution of nanotubes or nanofibers. As can be seen in Fig. 7(a), MWCNTs synthesized at 500 °C have diameters in the range 8–11 nm, at 600 °C have diameters in the range 13–16 nm, at 700 °C the diameter distribution further increased to 13–37 nm. It is reported that the diameter of these produced MWCNTs strongly depends on the size of the supported Co particles.27 A larger diameter of MWCNTs implies that a bigger diameter of Co particles formed during the reaction. These big particles can easily form at higher temperatures by Ostwald ripening. As can be seen in Fig. 7(d), when the temperature reached to 800 °C, the catalyst particles keep growing but are unable to support MWCNT growth, thus CNFs and amorphous carbon were dominated as the synthesized products, which often deactivate catalysts, as previously mentioned.
 |
| | Fig. 7
SEM images of MWCNTs formed by ethanol decomposition over Co(30 mol%)/Al2O3 catalyst at different temperatures: (a) 500, (b) 600, (c) 700, and (d) 800 °C. Insert figures show the distributions of external diameters of CNTs or CNFs. | |
From the SEM result, one can not decide which temperature is the best for ethanol decomposition to grow high-quality MWCNTs (fewer impurities, better degree of graphitization, and smoother tube-wall with few defects). Therefore, it is necessary to use TEM measurement to investigate the structure of MWCNT synthesized. Fig. 8 shows TEM images of MWCNTs formed by ethanol decomposition over Co(30 mol%)/Al2O3 catalyst at 500, 600 and 700 °C. From the TEM result it can be clearly seen that the structure of MWCNTs changed obviously when rising the reaction temperature. At 500 °C, fish-bone structure MWCNTs are the main products accompanied with a little amorphous carbon (Fig. 8(a) and (b)). All the carbon layers are angled at 40–45° to the axis of the tubes, indicating the defect of MWCNTs. When the temperature increased to 600 °C, the carbon layers of MWCNTs formed are more parallel to the tube axis than those formed at 500 °C, and the fringes of the walls for MWCNTs are relatively more perfect than those at 500 °C. For the MWCNTs synthesized at 700 °C, although perfect carbon layers can be formed (like the results obtained at 600 °C), a wider diameter distribution suggests that diameter-controlled growth of MWCNTs could hardly be realized at this temperature. According to the results obtained above, it can be concluded that a temperature of 600 °C could accelerate ethanol decomposition effectively to grow high-quality MWCNTs when compared with other temperatures. Two main forms of CNTs are the parallel and fish-bone type. By control the experimental condition, the structure of CNTs could be transformed between the two types. For example, Bao et al. have reported that the CNT microstructural transformation from the fish-bone into highly crystalline tubular structure can be realized by adding an external electric field.18 Besides the reported Bao method, variations in the choice of the metal, the reaction temperature and time can also alter the ordering of the graphite planes from fish-bone to parallel.28–30 Zheng et al. have recently reported that the geometry of catalyst particles could affected by the reaction temperature during ethanol decomposition. A high temperature of 600 °C is more beneficial to the formation of Co nanoclusters with a highly elongated shape than the low temperature.31 And it has been reported that such a shape facilitates the synthesis of tubular structure with the graphene sheets aligned parallel to the tube axis.32 In our research work, a change of fish-bone tubes to straight tubes occurred when only the reaction temperature increased. According to the reported results, it can be speculated that the transformation is attributed to the change of the catalyst shape, which was influenced by the reaction temperature.
 |
| | Fig. 8
TEM images of MWCNTs formed by ethanol decomposition over Co(30 mol%)/Al2O3 catalyst at different temperatures: (a) 500, (c) 600, and (e) 700 °C. (b) (d) and (f) are the HRTEM images of single MWCNT in (a), (c), and (e), respectively. | |
At the optimum temperature for MWCNT growth, we investigated the influence of Co loadings on the morphology of carbon produced (Fig. 9). As can be seen in Fig. 9(a), a few MWCNTs formed on Co(10 mol%)/Al2O3, indicating the insufficient of active sites for catalytic decomposition of ethanol to synthesize MWCNTs. However, when the loading amounts further increased, no obvious differences in the density and diameter of MWCNTs are observed in Fig. 9(b)–(h) for these catalysts with different Co loadings (SEM image of MWCNTs synthesized over Co(30 mol%)/Al2O3 is displayed in Fig. 7(b), not shown here). The result suggests that the size of Co metal particles at 600 °C might change slightly when Co loadings were no less than 20 mol%. Investigation of the Co loadings on the deposited carbon yield is shown in Fig. 10. Here the deposited carbon yield is calculated according to eqn (2).
| |  | (2) |
 |
| | Fig. 9
SEM images of carbon products formed by ethanol decomposition over Co/Al2O3 catalysts with different Co loadings at 600 °C: (a) 10, (b) 20, (c) 40, (d) 50, (e) 60, (f) 70, (g) 80, and (h) 90 mol%. | |
 |
| | Fig. 10 Change in the carbon deposition yield as a function of Co loadings in Co/Al2O3 catalysts at 600 °C. | |
It indicates that the Co loadings mainly influence the yield of carbon produced. The yield increased gradually with Co loadings and reached a maximum value 80% for the Co(90 mol%)/Al2O3 catalyst.
During the reaction, a trace amount of acetaldehyde was detected, which suggests ethanol was decomposed by catalytic dehydrogenation over Co catalysts. However, the amount of acetaldehyde generated is less than 1%. Because of the long time of the experiments and the small amount of liquid product, the reaction effluents were condensed to improve the accuracy of the analyses of the gaseous products. Fig. 11 shows a GC/FID chromatogram which indicating the characteristic peaks of the gaseous products of an effluent gas sample collected randomly during the reaction. It can be seen that four peaks at 0.4, 1.9, 4.9 and 11.9, attributed to H2, CO, CH4 and CO2, respectively, were observed from the chromatogram. The order of fraction of various gas in the effluent gas was H2 > CO > CH4 > CO2. On the basis of the obtained products, a mechanism for the surface reaction steps can be proposed, which is shown in Fig. 12. Additionally, after we systemically investigated the variation of gaseous products, it was found that the amount of H2 and CO increased while CH4 and CO2 decreased as the reaction temperature rises. The following reactions may describe the changes of gaseous products with temperatures in our reaction system.| | | CH3CH2OH → CO + H2 + CH4, ΔH = 50 kJ mol−1 | (3) |
| | | CH4 → C + 2H2, ΔH = 75 kJ mol−1 | (4) |
| | | 2CO → C + CO2, ΔH = −171 kJ mol−1 | (5) |
 |
| | Fig. 11
GC/FID
chromatogram of a gas sample collected randomly during the reaction. | |
 |
| | Fig. 12 Proposed surface decomposition reactions of ethanol. | |
The first reaction is the most endothermic one and it is thermodynamically favorable, so the high temperature favors the production of H2 and CO. Therefore, the higher the temperature the larger the amount of H2 and CO can be obtained. Eqn (4) is also a thermodynamically favorable reaction, thus the synthesized CH4 in eqn (3) will partially decomposed to produce carbon and hydrogen. This is the reason that the amount of CH4 decreased when the reaction temperature increased. The most exothermic reaction is the Boudouard reaction (eqn (5)) and it is favored at low temperatures, which lead to the decrease of the amount of CO2 when increasing the reaction temperature.
Reactions related to carbon formation are eqn (4) and (5), and carbon deposition will depend on these ones. The mechanism of CNT, CNF and amorphous carbon formation is mainly composed of the following steps.33 Firstly, ethanol adsorption on the surface of catalysts and the decomposition process occurred to produce carbon atoms. Secondly, the surface carbon atoms dissolve into the cobalt particles on the gas side, and diffuse through cobalt. Finally, carbon precipitated on the rare side of the cobalt particle to form CNTs, CNFs or amorphous carbon. The types of carbon products synthesized were determined by the difference between the carbon formation rate and carbon diffusion rate.
4. Conclusions
Ethanol
decomposition over Co/Al2O3 catalysts with different Co loadings was investigated in the temperature range 500–800 °C. The influence of the reaction temperature and loading amounts on MWCNT growth and hydrogen production was discussed systematically. Results in this work show that catalytic decomposition of ethanol can be carried out at a relatively low temperature (< 800 °C) to co-produce hydrogen and MWCNTs. The hydrogen yield increased with temperature rising from 500 to 700 °C, but when considering hydrogen production and high-quality MWCNT growth at the same time, 600 °C is the optimum temperature for ethanol decomposition over 20–90 mol% Co/Al2O3 catalysts. According to the gaseous products synthesized, explanations on the reaction pathways of ethanol decomposition were suggested.
Acknowledgements
The project was supported by the financial supports of the International cooperation research program of National Natural Science Foundation of China (No. 21061130551), the National Basic Research Program of China (973 Program) (No. 2009CB626611), the Ph. D. Programs Foundation of Ministry of Education of China (No. 20096101110002), the National Natural Science Foundation of China (Nos. 20873099 and 10974152), and NWU Doctorate Dissertation of Excellence Funds (No. 09YYB04).
References
- J. D. Holladay, Y. Wang and E. Jones, Chem. Rev., 2004, 104, 4767 CrossRef CAS.
- A. Haryanto, S. Fernando, N. Murali and S. Adhikari, Energy Fuels, 2005, 19, 2098 CrossRef CAS.
- J. Wang, M. Jiang, Y. Yao, Y. Zhang and J. Cao, Fuel, 2009, 88, 1572 CrossRef CAS.
- G. Italiano, C. Espro, F. Arena, A. Parmaliana and F. Frusteri, Appl. Catal., A, 2009, 365, 122 CrossRef CAS.
- D. P. Serrano, J. A. Botas, J. L. G. Fierro, R. G. Lopez, P. Pizarro and G. Gomez, Fuel, 2010, 89, 1241 CrossRef CAS.
- M. Manzoli, A. Chiorino and F. Boccuzzi, Appl. Catal. B, 2005, 57, 201 CrossRef CAS.
- S. Rabe and F. Vogel, Appl. Catal., B, 2008, 84, 827 CrossRef CAS.
- X. Chen, K. Honda and Z. G. Zhang, Appl. Catal., A, 2005, 288, 86 CrossRef CAS.
- N. Laosiripojana, W. Sutthisripok and S. Assabumrungrat, Chem. Eng. J., 2007, 127, 31 CrossRef CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. Heer, Science, 2002, 297, 787 CrossRef CAS.
- R. G. Ding, G. Q. Lu, Z. F. Yan and M. A. Wilson, J. Nanosci. Nanotechnol., 2001, 1, 7 CrossRef CAS.
- J. Qiu, Z. Wang, Z. Zhao and T. Wang, Fuel, 2007, 86, 282 CrossRef CAS.
- A. Bachmatiuk, F. Borrnert, F. Schaffel, M. Zaka, G. S. Martynkowa, D. Placha, R. Schonfelder, P. M. F. J. Costa, N. Ioannides, J. H. Warner, R. Klingeler, B. Buchner and M. H. Rummeli, Carbon, 2010, 48, 3175 CrossRef CAS.
- Y. Wang, Y. Q. Liu, D. C. Wei, L. C. Cao, L. Fu, X. L. Li, H. Kajiura, Y. M. Li and K. Noda, J. Mater. Chem., 2007, 17, 357 RSC.
- H. Qi, C. Qian and J. Liu, Chem. Mater., 2006, 18, 5691 CrossRef CAS.
- C. Pan and Q. Bao, J. Mater. Sci. Lett., 2002, 21, 1927–1929 CrossRef CAS.
- Q. Bao and C. Pan, Nano Technol., 2006, 17, 1016–1021 CrossRef CAS.
- Q. Bao, H. Zhang and C. Pan, Appl. Phys. Lett., 2006, 89, 063124 CrossRef.
- W. Li, H. Wang, Z. Ren, G. Wang and J. Bai, Appl. Catal., B, 2008, 84, 433 CrossRef CAS.
- G. Marban, A. Lopez, I. Lopez and T. V. Solis, Appl. Catal., B, 2010, 99, 257 CrossRef CAS.
- U. Narkiewice, M. Podsiadly, R. Jedrzejewski and I. Pelech, Appl. Catal., A, 2010, 384, 27 CrossRef.
- M. S. Batista, R. K. S. Santos, E. M. Assaf, J. M. Assaf and E. A. Ticianelli, J. Power Sources, 2004, 134, 27 CrossRef CAS.
- J. Su, Q. Liu, Z. Liu and Z. Huang, Ind. Eng. Chem. Res., 2008, 47, 4295 CrossRef CAS.
- J. I. Villacampa, C. Royo, E. Romeo, J. A. Montoya, P. D. Angel and A. Monzon, Appl. Catal., A, 2003, 252, 363 CrossRef CAS.
- R. M. Almeida, H. V. Fajardo, D. Z. Mezalira, G. B. Nuernberg, L. K. Noda, L. F. D. Probst and N. L. V. Carreno, J. Mol. Catal. A: Chem., 2006, 259, 328 CrossRef.
- T. Tsoufis, P. Xidas, L. Jankovic, D. Gournis, A. Saranti, T. Bakas and M. A. Karakassides, Diamond Relat. Mater., 2007, 16, 155 CrossRef CAS.
- K. Y. Tran, B. Heinrichs, J. F. Colomer, J. P. Pirard and S. Lambert, Appl. Catal., A, 2007, 318, 63 CrossRef CAS.
- W. Qian, T. Liu, Z. Wang, H. Yu, Z. Li, F. Wei and G. Luo, Carbon, 2003, 41, 2487 CrossRef CAS.
- S. L. Fiedler, S. Izvekov and A. Violi, Carbon, 2007, 45, 1786 CrossRef CAS.
- Y. Sawant, R. S. Somani and H. C. Bajaj, Carbon, 2010, 48, 668 CrossRef.
- L. Zheng, X. Liao and Y. T. Zhu, Mater. Lett., 2006, 60, 1968 CrossRef CAS.
- S. Helveg, C. L. Cartes, J. Sehested, P. L. Hansen, B. S. Clausen, J. R. R. Nielsen, F. A. Pedersen and J. K. Norskov, Nature, 2004, 427, 426 CrossRef CAS.
- D. Chen, K. O. Christensen, E. O. Fernandez, Z. Yu, B. Totdal, N. Latorre, A. Monzon and A. Holmen, J. Catal., 2005, 229, 82 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 

