Dual role of ionic liquids as phase transfer catalyst and solvent for glycosidation reactions†
Vineet
Kumar‡
,
Ian
Jamie Talisman‡
,
Omar
Bukhari
,
Jacqueline
Razzaghy
and
Sanjay V.
Malhotra
*
Laboratory of Synthetic Chemistry, SAIC-Frederick Inc., National Cancer Institute at Frederick, 1050 Boyles Street, Frederick, MD-21702. E-mail: malhotrasa@mail.nih.gov; Fax: +1-91-301-846-5206; Tel: +1-301-846-5141
First published on 14th October 2011
Abstract
This report describes the dual role of ionic liquids as phase transfer catalysts and reaction media in heterogeneous glycosidation reactions. Thorough study using a diverse set of ionic liquids provided insight into the relationship between ionic liquid structure and catalytic activity in these reactions. For example, glycosidation was efficient in ionic liquid 1-hexyl-3-methylimidazolium hexafluorophosphate (HxMIm.PF6), and the O- and S-glycosides were produced exclusively in moderate to good yields. As an outcome of the preliminary screen, a tailored novel ionic liquid, 1-hydroxyhexyl-3-methylimidazolium hexafluorophosphate (HOHxMIm.PF6) was rationally designed to be immiscible with water and traditional organic solvents. This provided an advantage in ionic liquid recycling and product recovery via convenient triphasic extraction. The versatility of this methodology was demonstrated through glycosidation reaction on a wide variety of substrates including phenols (17–79%), thiophenols (24–97%), chalcone (44%), and flavone (50–67%). Furthermore, this study shows that the ionic liquid could be employed for at least three runs without apparent loss in activity.
Introduction
Aromatic O-glycosides are frequently isolated as natural product extracts from traditional herbal medicines. Biologically active examples of these compounds include hyuganosides, which are carbohydrate linked benzofurans that demonstrate hepatoprotective activity;1,2 myricanenes A and B, which consist of glycosidated polyhydroxylated biphenyl rings and exhibit anti-allergic effects;3 and rutin, a flavanoid glycoside known as a potent VEGF inhibitor.4 Similarly glycosylated polyphenolic and carboline compounds include analgesics and anticonvulsants,5 while other therapeutically valuable compounds, doxorubicin,6,7daunomycin,8,9 and etoposide,10,11 also contain an aromatic core which is remotely conjugated to a carbohydrate. The large chemical and biological diversity of natural aromatic glycosides, therefore, makes the synthetic development of these materials a worthwhile endeavor. Consequently, this warrants development of efficient methodology to construct aromatic O-glycosidic bonds selectively in high yields.Glycosyl halides tend to be the most frequently employed donors in aromatic glycosidations; they have been arylated in mixed methanol aqueous systems, aprotic solvents, and biphasic systems under phase transfer conditions.12 This last, heterogeneous technique was originally reported for the coupling of 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide with ammonium resin bound nitrophenoxides13 and for the coupling of 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl bromide with various phenols.14,15 The mechanism of glycosidation involved the transportation of phenoxide anion in the aqueous layer to the glycosyl halide in the organic layer by phase transfer catalysts (PTCs), such as tetrabutylammonium chloride (TBACl).
Ionic liquids (ILs) consist only of ions, are usually liquid at room temperature, and have emerged as high-tech solvents of the future.16 This is because of their advantageous properties; they dissolve a wide range of organic and inorganic compounds; they are useful in extractions and distillations because of their immiscibility with certain organic solvents;17–19 and they are easy to handle and are highly polar media that may accelerate reaction rates. Most important, their properties can be adjusted to the needs of the reaction by proper manipulation of cations and anions. As a consequence of these merits, ILs have been employed in a wide variety of reactions.20–22 Recently, there has been increasing interest in employing ILs in place of traditional PTCs23 in biphasic reactions. ILs containing imidazolium, phosphonium, ammonium, and pyridinium cations have been shown to act as phase transfers catalysts for fluorinations, nucleophilic substitutions, etherifications, and benzoin condenations.24–30 One particularly useful feature of ILs is their potential to be tailored for a particular task, and differences in cation structure and chain length have been shown to have a large impact on their effectiveness as PTCs.
We recently demonstrated that halide molten salts were effective media for O-glycosidation of phenols at elevated temperatures under ambient conditions.31 This methodology was extended for use in room temperature ILs (RTILs), where in situ generation of silver carbene complexes in ILs promoted O-glycosidation of a variety of acceptors. The advantages of ILs as PTCs and our interest in developing aromatic O-glycosides for the treatment of cancer motivated us to examine phase transfer catalyzed glycosidation using a variety of ILs. Herein, we report the efforts to understand the role of ILs in the phase transfer catalyzed glycosidation reaction of glycosyl halides and also novel ILs that can be efficiently used and recycled in these reactions.
Results and Discussion
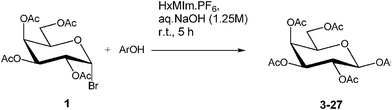
To demonstrate the proof-of-concept that ionic liquids are effective phase transfer catalysts in O-glycosidation reactions, we evaluated a set of ILs for the reaction of 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide (1) with 4-nitrophenol under biphasic conditions using aqueous sodium hydroxide and chloroform (Table 1). We examined the reaction with respect to the type of IL, stoichiometry of the IL, and temperature. The results clearly demonstrated the imidazolium halides such as 1-butyl-3-methyl imidazolium chloride worked effectively as PTCs for these reactions at 45 °C (Table 2).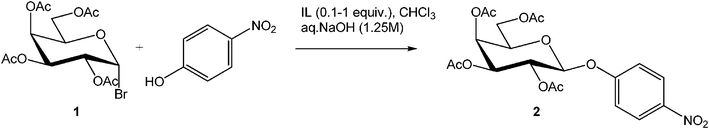
| Entry | Halide IL PTC | Time (h) | % Yield |
|---|---|---|---|
| a 1 (1 equiv.), 4-nitrophenol (2 equiv.) and IL (1 equiv.) were stirred in biphasic CHCl3 (2 mL) and 1.25 M NaOH (1.1 mL) at r.t. b Reaction was incomplete. | |||
| 1 | - | 48 | Trace |
| 2 | BMIm.Cl | 30 | 80 |
| 3 | BMIm.Br | 40 | 73 |
| 4 | BMIm.I | 40 | 40 |
| 5 | EMIm.Cl | 24 | 23 |
| 6 | HxMIm.Cl | 24 | 79 |
| 7 | OMIm.Cl | 24 | 37 |
| 8 | BMPr.Cl | 40 | 74 |
| 9 | BPy.Cl | 40 | 68 |
| 10 | BMIm.N(CN)2 | 30 | 66 |
| 11 | BMIm.BF4 | 30 | 83 |
| 12 | BMIm.OAc | 30 | 83 |
| 13 | BMIm.PF6 | 48 | 23b |
| 14 | BMIm.Tf2N | 48 | 34b |
| 15 | BMMIm.Cl | 30 | 65b |
| Entry | IL | Equiv. | Temp. (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| a 1 (1 equiv.), 4-nitrophenol (2 equiv.) and IL were stirred in biphasic CHCl3 (2 mL) and 1.25 M NaOH (1.1 mL). | |||||
| 1 | BMIm.Cl | 1.0 | 25 | 40 | 80 |
| 2 | BMIm.Cl | 1.0 | 45 | 6 | 72 |
| 3 | BMIm.Cl | 0.5 | 25 | 60 | 78 |
| 4 | BMIm.Cl | 0.5 | 45 | 7 | 75 |
| 5 | BMIm.Cl | 0.1 | 25 | 60 | 30 |
| 6 | BMIm.Cl | 0.1 | 45 | 7 | 60 |
| 7 | BMIm.Br | 1.0 | 25 | 18 | 70 |
| 8 | BMIm.Br | 0.5 | 45 | 27 | 74 |
| 9 | BMIm.I | 1.0 | 25 | 40 | 40 |
| 10 | BMIm.I | 0.5 | 45 | 27 | 49 |
| 11 | TBACl | 0.5 | 45 | 24 | 72 |
| 12 | TBABr | 0.5 | 45 | 24 | 54 |
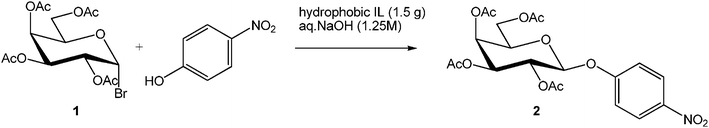
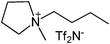
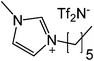
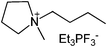
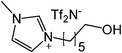
Our next goal was to explore the potential of RTILs to act both as PTCs and reaction media in our model glycosidation reaction of 1 with 4-nitophenol. Reports of ILs as both PTCs and also reaction media are relatively limited.28 Therefore, we screened a series of ILs containing imidazolium, piperidinium, pyridinium, morpholinium, uronium, thiouronium, and pyrrolidinium cations with several anions, such as bis(trifluoromethylsulfonyl)amide (Tf2N−), triethyltrifluorophosphate (Et3PF3−), and PF6−. The cations and anions were selected such that the ILs were immiscible with the aqueous phase giving a biphasic mixture when mixed with water. In general, imidazolium and pyridinium salts worked better than RTILs containing uronium, thiouronium, pyrrolidium, morpholinium, piperidinium, and ammonium cations. RTILs containing PF6− produced the glycosides in higher yields than the corresponding RTILs containing Tf2N− (Table 3, compare entries 10–12 with 14, 15, and 17), and a C6 alkyl chain on imidazolium ILs was determined to be optimal (Table 3, entries 13–16). These results may suggest that the hexyl-substituted imidazolium IL possesses properly balanced water penetration and hydrophobicity properties to achieve optimum conversion to product.
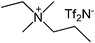
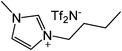
| Entry | IL | Time (h) | Yield | Entry | IL | Time (h) | Yield |
|---|---|---|---|---|---|---|---|
| a 1 (1 equiv.), 4-nitrophenol (2 equiv), and 1.25 M NaOH (1.1 mL) were stirred with IL (1.5 g) at r.t. | |||||||
| 1 |
|
18 | 35 | 10 |
|
18 | 42 |
| 2 |
|
18 | 35 | 11 |
|
18 | 34 |
| 3 |
|
18 | 23 | 12 |
|
4 | 36 |
| 4 |
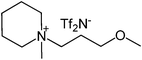
|
18 | 37 | 13 |
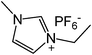
|
18 | 24 |
| 5 |
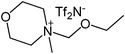
|
18 | 38 | 14 |
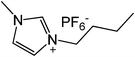
|
18 | 52 |
| 6 |
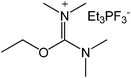
|
18 | 20 | 15 |
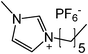
|
18 | 61 |
| 7 |
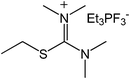
|
18 | 23 | 16 |
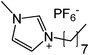
|
18 | 46 |
| 8 |
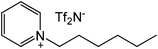
|
18 | 41 | 17 |
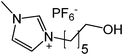
|
2 | 42 |
| 9 |
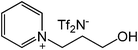
|
4 | 43 |
As the first screen showed that 1-hexyl-3-methylimidazolium hexafluorophosphate (HxMIm.PF6) gave the best result, we then evaluated the scope of the reaction using it as both solvent and PTC. As shown in Table 4 (entries 3–6, 24), electron deficient phenols substituted at the two or three position gave better yields than neutral or electron rich phenols. In general, moderate to good yields were obtained that were comparable to those reported in the literature using a biphasic water/chloroform system with ammonium PTCs.15 We also screened a series of thiophenols as acceptors (Table 5). In general, very high yields were obtained for both electron rich and electron deficient thiophenols, and the products were isolated exclusively as the β-anomers. In the case of 2-mercaptophenol, only the S-glycoside 35 was observed because of the higher nucleophilicity of the -SH over the -OH group. To the best of our knowledge this is the first report where S-glycosidation has been carried out using an ionic liquid medium.
| Entry | Phenol | Product | Yield | Entry | Phenol | Product | Yield |
|---|---|---|---|---|---|---|---|
| a 1 (1 equiv.), phenol (2 equiv), and 1.25 M NaOH (1.1 mL) were stirred with HxMIm.PF6 (1.5 g) at r.t. b Reaction done using BMIm.Cl/CHCl3/aqNaOH. | |||||||
| 1 | H | 3 | 54 | 14 | 4-OMe | 16 | 41 |
| 2 | 2-napthol | 4 | 42 | 15 | 4-Et | 17 | 54 |
| 3 | 2-CHO | 5 | 79 | 16 | 4-propyl | 18 | 40 |
| 4 | 3-NO2 | 6 | 70 | 17 | 4-sec-butyl | 19 | 48 |
| 5 | 3-CN | 7 | 76 | 18 | 4-butoxy | 20 | 55 |
| 6 | 3-Br | 8 | 65 | 19 | 4-phenoxy | 21 | 17 |
| 7 | 3-Cl | 9 | 59 | 20 | 4-benzyloxy | 22 | 35 |
| 8 | 3-OMe | 10 | 53 | 21 | 4-NAc b | 23 | 24 |
| 9 | 4-CN | 11 | 60 | 22 | 3,5-di Cl | 24 | 70 |
| 10 | 4-Cl | 12 | 53 | 23 | 4-OMe, 2-NO2 | 25 | 59 |
| 11 | 4-Br | 13 | 48 | 24 | 4-Cl, 3-Me | 26 | 66 |
| 12 | 4-I | 14 | 67 | 25 | 3,5-di OMe | 27 | 59 |
| 13 | 4-Ph | 15 | 48 |
| Entry | Acceptor | Product | % Yielda |
|---|---|---|---|
| a 1 (1 equiv.), thiophenol (2 equiv), and 1.25 M NaOH (1.1 mL) were stirred with HxMIm.PF6 (1.5 g) at r.t. | |||
| 1 | 4-isopropylbenzenethiol | 28 | 97 |
| 2 | 2-methoxybenzenethiol | 29 | 96 |
| 3 | 3-chlorobenzenethiol | 30 | 85 |
| 4 | 4-nitrobenzenethiol | 31 | 73 |
| 5 | 2-napthelenthiol | 32 | 66 |
| 6 | 4-methylbenzenethiol | 33 | 26 |
| 7 | 4-aminothiophenol | 34 | 24 |
| 8 | 2-mercaptophenol | 35 | 97 |
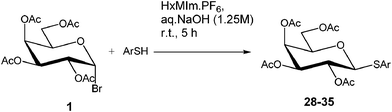
While we identified HxMIm.PF6 to be the best IL that played a dual role of PTC and solvent, this RTIL, due to high miscibility, possesses the challenge of separation from organic solvents such as dichloromethane and ethyl acetate (Fig. 1a). This makes recovery of the IL tedious viacolumn chromatography. We therefore attempted to facilitate the work-up procedure by modifying the IL structure. Hydroxylated ILs, such as 1-hydroxyethyl-3-methylimidazolium hexafluorophosphate (HOEMIm.PF6), have been described as “hyperpolar ILs” having polarities close to water.32 Also, it has been reported that these ILs have unique solubility properties and limited miscibility with dichloromethane.33 We speculated that introduction of a hydroxyl group onto the imidazolium ILs having longer side chains (C3-C6) would increase the polarity of the IL and decrease its miscibility with dichloromethane while retaining water immiscibility. Therefore, next we examined the C6-alkylated RTILs bearing a hydroxyl moiety in our model reaction. To the best of our knowledge, this is the first report of the synthesis of 1-hydroxyhexyl-3-methylimidazolium hexafluorophosphate (HOHxMIm.PF6, Table 3, entry 17), which was prepared by treatment of 1-hydroxyhexyl-3-methylimidazolium chloride34 with potassium hexafluorophosphate in water at room temperature. Interestingly, the glycosidation rate in hydroxylated RTILs was significantly improved in both the pyridinium and imidazolium series. Much to our delight, these RTILs exhibited limited miscibility with dichloromethane (Table 3, entries 9, 12, and 17) and water. Thus, employing novel HOHxMIm.PF6 facilitated easy RTIL recycling via triphasic separation of the aqueous layer, organic layer (product), and ionic liquid (Fig. 1b). It is worth mentioning that we did not observe any glycosidation of HOHxMIm.PF6 during the course of the reaction. As equimolar quantities of sodium hydroxide and phenol were used, it is likely that the base reacts exclusively with phenol due to its higher acidity than HOHxMIm.PF6.
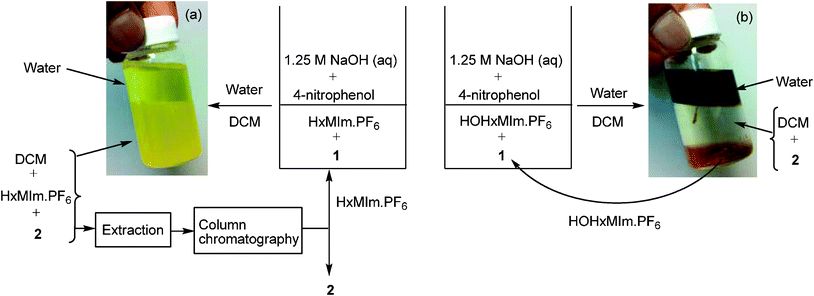 | ||
| Fig. 1 (a) A biphasic mixture of water (top layer), HxMIm.PF6 (lower layer), and dichloromethane (lower layer) during workup. (b) A triphasic mixture of water (top layer), dichloromethane (middle layer) and HOHxMIm.PF6 (bottom layer) during workup. | ||
The recycling studies were done on the model reaction of 1 and 4-nitrophenol using HxMIm.PF6 and HOHxMIm.PF6 (Fig. 1). In case of HxMIm.PF6, dichloromethane was added to the reaction mixture after completion to obtain a bilayer mixture (Fig. 1a). Both the product and HxMIm.PF6 were extracted into dichloromethane. After concentration in vacuo, the residue was subjected to silica gel chromatography, and pure product was obtained using a hexanes/ethyl acetate system (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30). HxMIm.PF6 was subsequently recovered by eluting with 10% methanol/dichloromethane. In the case of HOHxMIm.PF6, dichloromethane was added to the reaction mixture upon completion to obtain a tri-layer mixture (Fig. 1b). HOHxMIm.PF6 was recovered as the bottom layer, while the product was recovered from middle dichloromethane layer. In both the cases, recovered RTILs were recycled up to two times with no apparent loss of activity (Table 6).
:30). HxMIm.PF6 was subsequently recovered by eluting with 10% methanol/dichloromethane. In the case of HOHxMIm.PF6, dichloromethane was added to the reaction mixture upon completion to obtain a tri-layer mixture (Fig. 1b). HOHxMIm.PF6 was recovered as the bottom layer, while the product was recovered from middle dichloromethane layer. In both the cases, recovered RTILs were recycled up to two times with no apparent loss of activity (Table 6).
| Entry | Cycle | % Yield | |
|---|---|---|---|
| HxMIm.PF6 | HOHxMIm.PF6 | ||
| a 1 (1 equiv.), 4-nitrophenol (2 equiv), and 1.25 M NaOH (1.1 mL) were stirred with IL (1.5 g) at r.t. for 5 h. | |||
| 1 | 0 | 61 | 46 |
| 2 | 1 | 49 | 45 |
| 3 | 2 | 42 | 45 |
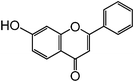
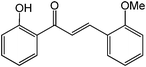
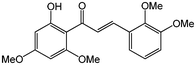
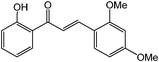
Chalcones are 1,3-diphenylprop-2-ene-1-ones that have exhibited valuable biological activity for various disease states. In particular, chalcones have been shown to disrupt cell cycles, inhibit blood vessel growth, and induce apoptosis. They possess cytotoxic activity in a variety of cell lines including B16 murine melanoma, HCT 116 human colon cancer, and A31 human epidermoid carcinoma.35,36 We evaluated the glycosidation of 2’-hydroxchalcone and another polyphenolic, 7-hydroxyflavone with 1 using HOHxMIm.PF6, HOHxMIm.Tf2N, HxMIm.PF6, and HxMIm.Tf2N (Table 7). In general, the PF6 counterion gave higher yields than Tf2N. Subsequently, a set of chalcone glycosides was synthesized in modest yields with complete β selectivity (Table 8).
A proposed mechanism of action for the IL as PTC and as reaction medium is given in Fig. 2. Given the limited solubility of hydrophobic ILs in water that has been reported in the literature,37 we propose initial migration of small quantities of the BMIm.PF6 into aqueous base, which contains the phenoxide ion. The imidazolium cation can subsequently coordinate to the phenoxide ion and increase the solubility of the anion in the IL. We speculate that after migration of phenoxide to the IL phase, in situ generated 1-butyl-3-methylimidazolium 4-nitrophenoxide (BMIm.PNP, 42, Fig. 2) promotes glycosidation of acceptor with glycosyl bromide. Subsequently, BMIm.Br may return to the aqueous phase to repeat the cycle. To support the proposed mechanism, we synthesized 42 using a reported method38 and treated it with 1 using BMIm.PF6 and aqueous NaOH (1.25 M) as biphasic reaction medium. The reaction was completed in 3 h to afford the product in 50% yield which implicates 42 as an active intermediate for the reactions.
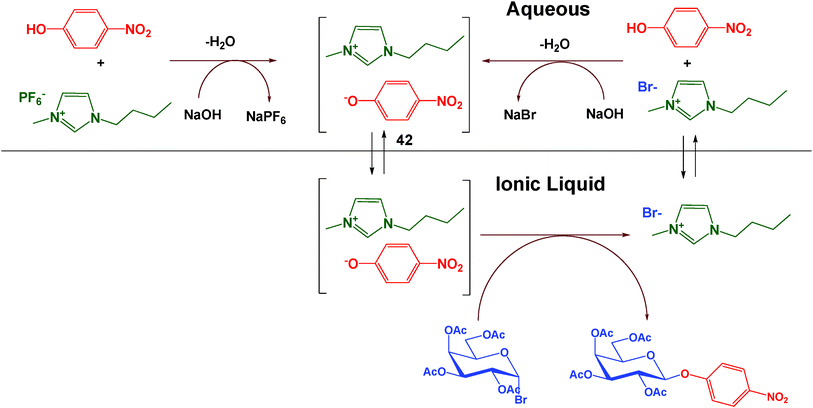 | ||
| Fig. 2 Proposed mechanism of action of dual-role ILs as PTCs and reaction media in O-glycosidation of 4-nitrophenol with 1. | ||
Conclusion
In summary, we have successfully demonstrated the application of ILs as PTCs in base-catalyzed glycosidation reactions. Hydrophobic RTILs have shown dual capabilities as PTCs and reaction media. The scope of the reaction was demonstrated with a variety of acceptor substrates e.g.phenols, thiophenols and chalcones. RTILs with a hydroxyl group in the cation elevated the reaction rate significantly and afforded a cleaner, triphasic work-up due to the immiscibility of these ILs with dichloromethane. This study has led to a novel ionic liquid i.e.HOHxMIm.PF6 that was tailored to improve the reaction rate and workup protocol. The RTILs were easily recovered and reused for multiple reaction cycles.Experimental
1-Hydroxyhexyl-3-methylimidazolium hexafluorophosphate (HOHxMIm.PF6): To a solution of 1-hydroxyhexyl-3-methylimidazolium chloride34 (15.5 g, 0.72 mol) in 100 mL water in an 250 mL erlenmeyer flask, potassium hexafluorophosphate (16 g, 0.86 mol) was added and the mixture was stirred for 2 h at room temperature. The formation of product was observed by appearance of a separate layer in the reaction mixture. The product was separated from the mixture using a separating funnel and washed with distilled water (3 × 30 mL) to afford the desired RTIL (12.7 g, 81%); 1H NMR (400 MHz, CD3OD) δ 8.74 (s, 1H), 7.56 (t, J = 1.8 Hz, 1H), 7.50 (t, J = 1.8 Hz, 1H), 4.18 (t, J = 7.4 Hz, 2H), 3.90 (s, 3H), 3.54 (t, J = 6.5 Hz, 2H), 1.89 (dt, J = 14.7, 7.5 Hz, 2H), 1.58 – 1.49 (m, 2H), 1.46 – 1.32 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 137.65, 124.79, 123.46, 62.69, 50.67, 36.36, 33.19, 30.93, 26.88, 26.20. LC-MS (ESI-TOF, positive mode) m/z calcd for [C10H19N2O]+ 183.1492; found 183.1490; LC-MS (ESI-TOF, negative mode) m/z calcd for [PF6]− 144.9636; found 144.9633.General procedure for the screen of ILs as PTCs in the biphasic glycosidation of 1 with 4-nitrophenol in chloroform
2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide (1, 1 equiv.), phenol (2 equiv.), and IL (as in Table 1 and 2) were combined with chloroform (2 mL) in a 14 mL borosilicate glass vial. Sodium hydroxide (1.1 mL, 1.25 M, 2 equiv.) was added and the resulting biphasic mixture was stirred until completion. To check the progress of the reaction, a small aliquot of reaction mixture was taken out and diluted with dichloromethane, and this solution was used for TLC spotting. The TLC was performed using an ethyl acetate/hexanes (40/60) solvent system as the mobile phase. After completion, the reaction mixture was diluted with 5 mL of dichloromethane. The aqueous and organic layers were separated, and the aqueous layer was washed with 3 × 25 mL of dichloromethane. The organics were combined, dried over sodium sulfate, and concentrated in vacuo. The resulting residue was loaded onto a silica gel cartridge for product isolation by flash chromatography using a gradient of ethyl acetate/hexanes (0–40% ethyl acetate). The desired product was obtained as white foam; the yields with corresponding RTILs are given in Table 1 and 2.General procedure for IL mediated glycosidation reactions
2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide (1, 1 equiv.) and phenol derivative (2 equiv.) were combined with IL (1.5 g) in a 14 mL borosilicate glass vial. Sodium hydroxide (1.1 mL, 1.25 M, 2 equiv.) was added and the resulting biphasic mixture was stirred at 25 °C until completion of the reaction. To check the progress of the reaction, a small aliquot of reaction mixture was taken out and diluted with dichloromethane, and this solution was used for TLC spotting. The TLC was performed using an ethyl acetate/hexanes (40/60) solvent system as the mobile phase. After completion of the reaction, the reaction mixture was diluted with 5 mL of dichloromethane. The aqueous and organic layers were separated, and the aqueous layer was washed with 3 × 25 mL of dichloromethane. The organics were combined, dried over sodium sulfate, and concentrated in vacuo. The resulting residue was loaded onto a silica gel cartridge for product isolation by flash chromatography using a gradient of ethyl acetate/hexanes (0–40% ethyl acetate). The desired product was obtained as white foam; the yields with corresponding times are given in Table 4.Recycling of HOHxMIm.PF6 from the reaction mixture
The ability to recycle HOHxMIm.PF6 was demonstrated via the reaction of 1 and 4-nitrophenol. Once the reaction was complete, water (5 mL) and dichloromethane (10 mL) were added to the reaction mixture. The resulting mixture formed a triphasic system. The IL was recovered from the bottom layer and the product was recovered from the middle dichloromethane layer. The top aqueous layer was discarded. The recovered IL was dried under vacuum at 80 °C overnight and used for next cycle without further purification; results are given in Table 4.Recycling of HxMIm.PF6 from the reaction mixture
The ability to recycle the ionic liquids was demonstrated via the reaction of 1 and 4-nitrophenol in HxMIm.PF6. Once the reaction was complete, water (5 mL) and dichloromethane (5 mL) were added to the reaction mixture. The organic and aqueous layers were separated and the residual product in the aqueous layer was extracted with dichloromethane (3 × 5 mL). The organic layers were combined, dried over sodium sulfate, and concentrated in vacuo. The residue was loaded onto a silica gel cartridge to isolate the product. The aqueous layer was concentrated in vacuo to recover the ionic liquid, which was dried under vacuum at 80 °C overnight and used for next cycle without further purification. The results are given in Table 6.Acknowledgements
The authors would like to thank the NCI Developmental Therapeutics Program. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E.References
- H. Matsuda, T. Murakami, T. Kageura, K. Ninomiya, I. Toguchida, N. Nishida and M. Yoshikawa, Bioorg. Med. Chem. Lett., 1998, 8, 2191–2196 CrossRef CAS.
- T. Morikawa, H. Matsuda, N. Nishida, T. Ohgushi and M. Yoshikawa, Chem. Pharm. Bull., 2004, 52, 1387–1390 CrossRef CAS.
- H. Matsuda, T. Morikawa, J. Tao, K. Ueda and M. Yoshikawa, Chem. Pharm. Bull., 2002, 50, 208–215 CrossRef CAS.
- R. Schindler and R. Mentlein, J. Nutr., 2006, 136, 1477–1482 CAS.
- H. Kikuchi, N. Takahashi and Y. Oshima, Tetrahedron Lett., 2004, 45, 367–370 CrossRef CAS.
- F. Arcamone, G. Cassinel, G. Fantini, A. Grein, P. Orezzi, C. Pol and C. Spalla, Biotechnol. Bioeng., 1969, 11, 1101–1110 CrossRef CAS.
- A. Dimarco, M. Gaetani and B. Scarpina, Cancer Chemoth. Rep. 1, 1969, 53, 33–37 CAS.
- A. Dimarco, L. Valentine, B. M. Scarpinato, T. Dasdia, M. Soldati, M. Gaetani, P. Orezzi and R. Silvestrini, Nature, 1964, 201, 706–707 CrossRef CAS.
- A. Dimarco, M. Gaetani, L. Dorigotti, M. Soldati and O. Bellini, Cancer Chemoth. Rep., 1964, 31–38 CAS.
- C. Kellerju, M. Kuhn, A. V. Wartburg and H. Stahelin, J. Med. Chem., 1971, 14, 936–940 CrossRef.
- G. L. Chen, L. Yang, T. C. Rowe, B. D. Halligan, K. M. Tewey and L. F. Liu, J. Biol. Chem., 1984, 259, 3560–3566 Search PubMed.
- M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266–1281 CrossRef CAS.
- D. Dess, H. P. Kleine, D. V. Weinberg, R. J. Kaufman and R. S. Sidhu, Synthesis, 1981, 883–885 CrossRef CAS.
- K. Brewster, J. M. Harrison and T. D. Inch, Tetrahedron Lett., 1979, 5051–5054 CrossRef CAS.
- H. P. Kleine, D. V. Weinberg, R. J. Kaufman and R. S. Sidhu, Carbohydr. Res., 1985, 142, 333–337 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- H. Zhao, S. Q. Xia and P. S. Ma, J. Chem. Technol. Biotechnol., 2005, 80, 1089–1096 CrossRef CAS.
- C. F. Poole and S. K. Poole, J. Chromatogr., A, 2010, 1217, 2268–2286 CrossRef CAS.
- Z. G. Lei, C. Y. Li and B. H. Chen, Sep. Purif. Rev., 2003, 32, 121–213 CrossRef CAS.
- N. Jain, A. Kumar, S. Chauhan and S. M. S. Chauhan, Tetrahedron, 2005, 61, 1015–1060 CrossRef CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- S. Toma, M. Meciarova and R. Sebesta, Eur. J. Org. Chem., 2009, 321–327 CrossRef CAS.
- C. M. Starks, J. Am. Chem. Soc., 1971, 93, 195–199 CrossRef CAS.
- S. S. Shinde, H. M. Chi, B. S. Lee and D. Y. Chi, Tetrahedron Lett., 2009, 50, 6654–6657 CrossRef CAS.
- J. Bender, D. Jepkens and H. Huskent, Org. Process Res. Dev., 2010, 14, 716–721 CrossRef CAS.
- A. Perosa, P. Tundo, M. Selva, S. Zinovyev and A. Testa, Org. Biomol. Chem., 2004, 2, 2249–2252 CAS.
- G. D. Yadav and S. P. Tekale, Org. Process Res. Dev., 2010, 14, 722–727 CrossRef CAS.
- N. M. T. Lourenco and C. A. M. Afonso, Tetrahedron, 2003, 59, 789–794 CrossRef CAS.
- L. W. Xu, Y. Gao, J. J. Yin, L. Li and C. G. Xia, Tetrahedron Lett., 2005, 46, 5317–5320 CrossRef CAS.
- D. W. Kim, C. E. Song and D. Y. Chi, J. Am. Chem. Soc., 2002, 124, 10278–10279 CrossRef CAS.
- V. Kumar, I. J. Talisman and S. V. Malhotra, Eur. J. Org. Chem., 2010, 3377–3381 CrossRef CAS.
- S. G. Zhang, X. J. Qi, X. Y. Ma, L. J. Lu and Y. Q. Deng, J. Phys. Chem. B, 2010, 114, 3912–3920 CrossRef CAS.
- V. Najdanovic-Visak, A. Rodriguez, Z. P. Visak, J. N. Rosa, C. A. M. Afonso, M. N. da Ponte and L. P. N. Rebelo, Fluid Phase Equilib., 2007, 254, 35–41 CrossRef CAS.
- K. Shimojo, K. Nakashima, N. Kamiya and M. Goto, Biomacromolecules, 2006, 7, 2–5 CrossRef CAS.
- M. L. Go, X. Wu and X. L. Liu, Curr. Med. Chem., 2005, 12, 483–499 CAS.
- R. De Vincenzo, C. Ferlini, M. Distefano, C. Gaggini, A. Riva, E. Bombardelli, P. Morazzoni, P. Valenti, F. Belluti, F. O. Ranelletti, S. Mancuso and G. Scambia, Cancer Chemother. Pharmacol., 2000, 46, 305–312 CrossRef CAS.
- D. S. H. Wong, J. P. Chen, J. M. Chang and C. H. Chou, Fluid Phase Equilib., 2002, 194, 1089–1095 CrossRef.
- S. Katsuta, K. Imai, Y. Kudo, Y. Takeda, H. Seki and M. Nakakoshi, J. Chem. Eng. Data, 2008, 53, 1528–1532 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, GCOSY, GHSQC data/spectra, and HRMS data relating to compounds 5, 8–10, 12, 14, 15, 17–36, 38–42 and RTIL HOHxMIm.PF6. See DOI: 10.1039/c1ra00385b |
| ‡ Both authors have equal contribution in this work. |
| This journal is © The Royal Society of Chemistry 2011 |